Farmakologia opioidowych leków przeciwbólowych. Opioidowe leki przeciwbólowe. Rodzaje, mechanizm działania, zastosowanie Wskazania blokery receptorów opioidowych
Leczenie farmakologiczne alkoholizmu
Wszystkie rodzaje leków na uzależnienie od alkoholu, które są stosowane w walce z tym destrukcyjnym głodem, można podzielić na 3 klasy:
- Wywoływanie nietolerancji alkoholu
- Zmniejszenie pragnienia alkoholu
- Częściowe niwelowanie „zespołu odstawienia” (zespołu kaca)
Leki z trzeciej grupy, do których należą Alkazeltser, Medichronal i inne leki działające jak „narkotyki”, są uważane za leki przeciwalkoholowe tylko nominalnie, ponieważ choć rano poprawiają samopoczucie, wieczorem dodatkowo prowokują do niekontrolowanego picia. Do tego zalicza się także zwykła aspiryna czy paracetamol, preparaty multiwitaminowe oraz cytoflawina (lek ten jest połączeniem dużej dawki kwasu bursztynowego i witamin). Te środki oczywiście poprawiają zdrowie po kacu, ale w żaden sposób nie zwalczają uzależnienia od alkoholu.
Informacja zwrotna od pensjonariusza, który pomyślnie ukończył kurs rehabilitacyjny w ośrodku
Leki z drugiej grupy stanowią tzw. „złoty standard” w leczeniu alkoholizmu w Ameryce i Europie, jednak większość z nich nie jest zarejestrowana w krajach byłego Związku Radzieckiego. Poza tym wygórowana cena (od 100 USD) za kurs terapeutyczny jest dla przeciętnego Słowianina nie do przyjęcia. Domowy lek Proproten – 100 nie sprostał oczekiwaniom i stosuje się go najczęściej, gdy trzeba zaprzestać picia i złagodzić dolegliwości po wytrzeźwieniu.
Dlatego obecnie najczęściej stosowaną klasą leków są leki wywołujące nietolerancję napojów alkoholowych. Stosowane są w ramach tzw. „terapii awersyjnej”. Omówiono główne leki wymienione poniżej.
Trzecia klasa:
Disulfiram. Często nazywa się go również Teturammus lub Antabuse, Abstinil i innymi nazwami. Uważany jest za najbardziej znany i „zasłużony” narkotyk tej grupy. Działanie tego środka opiera się na blokowaniu niektórych enzymów odpowiedzialnych za rozkład alkoholu we krwi. W rezultacie alkohol, który pijesz, zamienia się w aldehyd octowy. Substancja ta jest niezwykle toksycznym związkiem, który powoduje nudności, wymioty, silne bóle głowy, ataki paniki, szybkie bicie serca i inne negatywne objawy. Pacjent przyjmujący Disulfiram po prostu nie może pić alkoholu, ponieważ po każdym kieliszku staje się on tak zły, że picie traci jakikolwiek efekt relaksujący.
Disulfiram był z powodzeniem stosowany przez krajowych narkologów w czasach Związku Radzieckiego, gdzie alkoholicy trafiający do przychodni lekarskich i porodowych byli zmuszeni zażywać lek razem z alkoholem, po czym pojawiły się wyżej opisane objawy. W rezultacie po kilkukrotnym powtórzeniu procedury, którą naukowo nazywa się „testem awersji”, z przychodni opuściła się osoba, u której nie tylko smak, ale nawet widok lub wzmianka o alkoholu wywoływała u niego chęć wymiotowania. Minusem było to, że odruch ten szybko zanikał bez okresowego „odświeżania” zabiegu. W rezultacie pacjent stanął w obliczu kolejnego objadania się i nowego kursu w specjalistycznej placówce.
Disulfiram wywołuje ciężką reakcję na alkohol, jednak terapię można prowadzić tylko w krótkich cyklach, ponieważ sam lek jest toksyczny i przy długotrwałym stosowaniu ma szkodliwy wpływ na wątrobę, wywołując zapalenie wątroby i układ nerwowy ( różne zapalenie wielonerwowe). Lek może również powodować psychozę. Wielu doświadczonych alkoholików wypróbowało już na sobie działanie disulfiramu i nie zgadza się na ponowne leczenie. Stosowanie leku bez wiedzy osoby uzależnionej jest trudne ze względu na wyraźny metaliczny smak, który można łatwo wyczuć w każdym produkcie spożywczym.
Esperala. Tak naprawdę to wciąż ten sam Disulfiram, tyle że produkowany przez francuskich producentów, co zapewnia lepszy stopień oczyszczenia, dzięki czemu występowanie skutków ubocznych jest rzadsze. W przeciwnym razie produkt jest analogiem oryginalnego disulfiramu.
Podskórne wstrzyknięcie „Esperalu” w postaci sterylnych tabletek jest powszechnie znany pod nazwą „filtr”. Zabieg ten wykonywany jest chirurgicznie na plecach lub brzuchu, w zależności od wymagań producenta. Następnie lek wchłania się stopniowo, zapewniając stałe utrzymanie wymaganego stężenia leku we krwi pacjenta. Implantacja odbywa się przy ścisłym przestrzeganiu środków sanitarnych i higienicznych; używane są jednorazowe instrumenty medyczne.
Implantacja „Esperala” Wykonywany jest w znieczuleniu miejscowym i nie powoduje dyskomfortu. Okres ważności tego środka może wynosić od trzech miesięcy do półtora roku.
Lidevin. Jest połączeniem disulfiramu i dwóch witamin (adeniny i nikotynamidu). Witaminy mają na celu przynajmniej częściowe zmniejszenie toksycznego działania disulfiramu system nerwowy. Lek jest lepiej tolerowany niż klasyczny disulfiram, ma jednak te same wady, co lek oryginalny.
Colma. Hiszpański produkt dostępny w formie roztworu do picia. Substancją czynną leku jest cyjanamid (nie mylić z cyjankiem), który ma działanie podobne do disulfiramu i powoduje nietolerancję alkoholu. Efekt występujący po zażyciu Colme jest nieco łagodniejszy niż reakcja na disulfiram, ale wystarczający, aby wywołać niechęć do alkoholu.
Produkt jest nietoksyczny i można go stosować aż do sześciu miesięcy bez szkody dla zdrowia. Lek nie ma zauważalnego koloru, smaku ani zapachu. Dzięki temu lekarze mogą go stosować bez wiedzy pacjenta. Wszystko to mogłoby uczynić Colme liderem leczenia „awersyjnego”. Jednak koszt leku jest wysoki. Opakowanie produktu, które wystarcza na kilka tygodni stosowania, kosztuje 40 dolarów, co jest nie do przyjęcia dla każdej rodziny, zwłaszcza biorąc pod uwagę mentalność alkoholików (tyle alkoholu można by za te pieniądze kupić!) .
Skuteczność i bezpieczeństwo leku Colme pozwala na włączenie go do wielu kursów leczenia alkoholizmu w Ameryce i Europie. Hiszpańska metoda leczenia uzależnień opiera się na leczeniu tym konkretnym narkotykiem.
Tetlong – 250. Jest to lek do podawania domięśniowego, który został opracowany przez doktora Sobetova. Lek to disulfiram o opóźnionym wchłanianiu. Po podaniu w tkankach tworzy się depozyt, który pozwala na utrzymanie stałego stężenia disulfiramu w organizmie. Kurs Tetlong jest lepiej tolerowany, ale lek ma wszystkie „problematyczne punkty” disulfiramu. Do zalet leku należy fakt, że Tetlong można stosować zarówno w leczeniu alkoholizmu, jak i narkomanii.
Pamiętaj, że stosowanie któregokolwiek z wymienionych leków jest możliwe wyłącznie zgodnie z zaleceniami i pod stałym nadzorem narkologa. Tylko specjalista może przepisać najbardziej optymalny lek w każdym konkretnym przypadku, wyjaśnić niuanse stosowania oraz monitorować i korygować postęp i efekt leczenia.
Nie zapominaj też, że nie ma „magicznej pigułki”, która raz na zawsze wyeliminuje ochotę na alkohol. Uzależnienie od alkoholu jest poważną chorobą i do jej leczenia należy podchodzić kompleksowo. Połączenie leków i technik, wsparte całkowitym wzajemnym zrozumieniem lekarza, pacjenta i bliskich, da oczekiwany rezultat.
Przegląd leków
Esperal to specjalistyczny lek stosowany w terapeutycznym leczeniu alkoholizmu. Działa na zasadzie powstrzymywania narkotyków. Środek ten ma zdolność hamowania enzymu dehydrogenazy aldehydu octowego, który automatycznie zwiększa stężenie aldehydu octowego. Lek powoduje trwałą negatywną reakcję na zapach i smak alkoholu. Esperal nie tylko eliminuje ochotę na alkohol, ale także radykalnie zmienia podejście pacjenta do napojów alkoholowych. Stosując stale Esperal, pacjent przestaje cieszyć się przyjemnymi doznaniami związanymi z alkoholem.
Tiamina– powinien być przepisywany wszystkim pacjentom zgłaszającym się do specjalisty w sprawie alkoholizmu. Głównym zadaniem tiaminy jest zapobieganie zespołowi Korsakowa i encefalopatii Gaye-Wernickego.
β-blokery– są przepisywane w celu łagodzenia objawów wegetatywnych. Z reguły w tym celu przepisywany jest propranolol lub atenolol. Leki te nie zapobiegają występowaniu drgawek i majaczenia, dlatego należy je przepisywać w połączeniu z innymi lekami.
Klonidyna– pobudza ośrodkowe receptory adrenergiczne. Klonidyna jest w stanie złagodzić objawy wegetatywne, w tym zwiększoną potliwość, nadciśnienie tętnicze, tachykardię i drżenie. Warto pamiętać, że lek nie ma wpływu na zapobieganie majaczeniu i drgawkom. Produkt ma działanie uspokajające, które potęguje zastosowanie środków uspokajających. Zaletami klonidyny jest brak trudności w oddychaniu i stan euforyczny.
Benzodiazepiny– główny lek w leczeniu alkoholowego zespołu odstawiennego. Zmniejszają prawdopodobieństwo wystąpienia drgawek i majaczenia, a także skracają czas trwania majaczenia, które już wystąpiło. Benzodiazepiny skutecznie zapobiegają napadom padaczkowym. Wadą jest możliwość kumulacji i zbyt silne działanie uspokajające. Wśród leków w tej grupie: Lorazepam, Oxazepam, Chlorazepat, Diazepam.
Karbamazepina– zalecany przy alkoholowym zespole odstawiennym i może być stosowany w celu uzyskania efektu terapeutycznego u pacjenta w trakcie leczenia łagodnego i umiarkowanego zespołu odstawienia. Nie wchodzi w interakcje z alkoholem, co pozwala na jego stosowanie nawet w przypadku obecności alkoholu we krwi. Jednorazowe zastosowanie jest skuteczne w przypadku łagodnego i umiarkowanego zespołu; w przypadku ciężkiego zespołu zaleca się również benzodiazepiny. Nie powoduje efektu euforii i nie powoduje ryzyka uzależnienia od leku.
Barbiturany– skuteczny środek łagodzący objawy odstawienia. Stosowanie barbituranów jest ograniczone ze względu na ich wysoką toksyczność i ryzyko uzależnienia. Możliwa depresja oddechowa i sercowa; powodować indukcję enzymów wątrobowych.
Tiapryd– lek przeciwpsychotyczny, który działa uspokajająco na pacjenta. Zaburzenia pozapiramidowe związane z jego stosowaniem są rzadkie, ponieważ działa on selektywnie na receptory D2-dopaminy. Produkt stosowany jest w Rosji, Francji i Niemczech w celu zwalczania objawów odstawienia. Sensowne jest przepisywanie tiaprydu w połączeniu z karbamazepiną i benzodiazepinami.
Haloperidol– wskazany przy występowaniu objawów psychopatologicznych – urojeniowych sądów, omamów, pobudzenia psychoruchowego. Lek należy łączyć z benzodiazepinami. Podczas prostego zespołu odstawiennego lek nie jest stosowany. Jego wadą jest ryzyko ostrej hiperkinezy, którą można leczyć diazepamem, difenhydraminą lub lekami antycholinergicznymi.
Proproten-100– jest obecnie najlepiej zbadanym lekiem przeciwciałowym. „Proproten-100” zawiera przeciwciała przeciwko białku S-100. Proproten badano na wszystkich poziomach struktur neuronalnych: komórkowym, synaptycznym, strukturalnym i ogólnoustrojowym. Lek wykazuje zrównoważony wpływ na stan psychiczny pacjentów. W zależności od pierwotnego stanu pacjenta lek ma zarówno działanie uspokajające, jak i stymulujące.
GHB– lek łagodzi objawy wegetatywne i ma dość wyraźne działanie uspokajające, ale może powodować prawdopodobieństwo halucynacji, co wiąże się z pośrednim stymulującym wpływem na neurony dopaminergiczne pacjenta.
Klometiazol– lek o silnym działaniu uspokajającym, stosowany w praktyce leczniczej od lat 20. ubiegłego wieku. Wykazuje dużą skuteczność w łagodzeniu objawów odstawienia. Stosuje się go wewnętrznie w postaci kapsułek lub roztworu. Przeciwwskazaniami do stosowania są obturacyjne choroby płuc i niewydolność oddechowa. Produkt używany wyłącznie na stałe.
Leki uczulające– stworzyć tzw. barierę chemiczną uniemożliwiającą picie alkoholu i wywołać u klienta poczucie lęku przed możliwymi przykrymi konsekwencjami spożycia alkoholu. Najpopularniejszym lekiem uczulającym stosowanym w leczeniu alkoholizmu jest teturam (disulfiram). Przepisywane wcześniej środki uczulające, metronidazol lub furazolidon, a także kwas nikotynowy, są obecnie rzadko przepisywane. Disulfiram jest przepisywany w leczeniu uzależnienia od alkoholu od połowy ubiegłego wieku.
Uważa się, że działanie terapeutyczne Teturamu wynika z obawy przed reakcją teturam-alkohol. Działanie tego produktu opiera się na blokowaniu specyficznego enzymu, w wyniku czego utlenianie alkoholu zatrzymuje się na etapie aldehydu octowego. Przed rozpoczęciem terapii lekiem Teturam pacjent jest informowany o potencjalnych konsekwencjach spożywania napojów alkoholowych.
Powszechną metodą terapii jest domięśniowe podawanie leku Esperal. Metodę tę stosuje się, gdy lekarze uznają, że inne metody leczenia są nieskuteczne. Wszczepiony lek jest stale wchłaniany do krwioobiegu, a jeśli pacjent wypije choćby odrobinę alkoholu, doświadcza poważnych objawów, a nawet tragicznego skutku.
Warunkiem terapii jest wysoki poziom zdrowia klienta, wysoka motywacja i systematyczne stosowanie leku. Disulfiramu nie należy przepisywać bez wiedzy klienta (mówimy o dodawaniu go do jedzenia lub napojów), gdyż skutki mogą być bardzo niebezpieczne.
Blokery receptorów opioidowych– to nowe leki opracowane specjalnie do leczenia uzależnienia od alkoholu. Tym samym odnotowano, że w mózgu funkcjonuje specyficzny endogenny układ opioidowy, który wytwarza substancje morfinopodobne (enkefaliny i endorfiny) wywołujące efekt euforyczny i przeciwbólowy.
Antagoniści opioidów blokują receptory, co pomaga zapobiegać przyjemnym odczuciom pojawiającym się po zażyciu narkotyków. Nie jest tajemnicą, że alkohol nie jest agonistą tych receptorów, jednak wiele jego skutków objawia się poprzez endogenny ośrodek opioidowy w ludzkim mózgu. Badania wykazały, że antagoniści receptorów opioidowych blokują działanie alkoholu.
Naltrekson zapobiega wzrostowi poziomu dopaminy spowodowanemu spożyciem alkoholu. Naukowcy odkryli, że efekt ten był zależny od dawki. Dopamina bierze bezpośredni udział we wzmacniającym działaniu alkoholu. Czas trwania remisji u pacjentów przyjmujących naltrekson jako lek dodatkowy był dłuższy niż u pacjentów przyjmujących placebo. Należy pamiętać, że naltrekson jest skuteczny jako lek przeciwnawrotowy, jeśli jest stosowany systematycznie przez okres 12 tygodni. Zdecydowanie zaleca się przepisywanie naltreksonu pacjentom z intensywnym, niekontrolowanym głodem alkoholu (piciem kompulsywnym).
Terapia lekowa powinna być połączona z wysoce zmotywowanym klientem. Skuteczność leczenia znacznie wzrasta w połączeniu ze wspomagającymi procedurami psychoterapeutycznymi. Lek nalmefen jest strukturalnie podobny do naltreksonu. Jednakże nalmefen nie jest hepatotoksyczny. Ponadto nalmefen jest uniwersalnym antagonistą receptorów opioidowych, który może blokować wszystkie typy receptorów opioidowych.
Akamprozat– na chwilę obecną naukowcy nie ustalili dokładnego mechanizmu działania tego leku. Naukowcy odkryli, że lek jest w stanie modulować aktywność receptorów glutaminianu i receptorów GABA. Przewlekłe zatrucie alkoholem powoduje zmniejszenie aktywności hamującego układu GABA i zwiększenie aktywności pobudzającego układu glutaminianowego alkoholika. Zmiany te pozostają niezmienione przez długi czas po zaprzestaniu picia alkoholu. Akamprozat jest strukturalnie podobny do GABA i jest zdolny do zwiększania aktywności układu GABA. Akamprozat osłabia działanie układu glutaminianowego, wpływając na receptory N-metylo-D-asparaginianu (NMDA) oraz kanały wapniowe. Pierwsze próby zastosowania leku w praktyce klinicznej rozpoczęły się we Francji w 1989 roku. W tej chwili lek jest zatwierdzony w wielu krajach świata (ponad 30). Łączna liczba leczonych pacjentów przekracza 1 milion. Badania wykazały, że akamprozat zmniejsza spożycie alkoholu w warunkach swobodnego dostępu, nie zmieniając przy tym zachowań żywieniowych, nie ma potencjału narkotycznego i nie wywiera innego działania farmakologicznego niż te, które pomagają ograniczyć spożycie alkoholu.
Środki serotoninergiczne– Związek serotoniny z alkoholem jest dość złożony. Uważa się, że osoby uzależnione próbują normalizować niski poziom hormonu serotoniny w mózgu za pomocą alkoholu. Udokumentowano, że serotonina bierze udział we wzmacniającym działaniu alkoholu. Ponadto niski poziom serotoniny stymuluje zachowania impulsywne, co prowadzi do spożywania napojów alkoholowych. Zaburzeniom metabolizmu serotoniny może towarzyszyć stan lękowy i depresja, w takim przypadku alkohol może być środkiem samoleczenia. Eksperci uważają, że inhibitory wychwytu zwrotnego serotoniny to leki serotoninergiczne, do których zaliczają się sertralina, fluoksetyna, fluwoksamina, citalopram. Ta grupa leków została opracowana w latach 80. ubiegłego wieku do leczenia zaburzeń depresyjnych. Działanie inhibitorów wychwytu zwrotnego serotoniny polega na tym, że blokują wychwyt zwrotny serotoniny, co powoduje wzrost poziomu serotoniny w szczelinie synaptycznej.
Inne leki. Stosowanie doksepiny zalecane przez część ekspertów w leczeniu zespołu odstawiennego można nazwać niedopuszczalnym ze względu na duże ryzyko ewentualnych powikłań. Chodzi o to, że możliwe jest niedociśnienie tętnicze, arytmia i toksyczne delirium. Stosowanie fenotiazynowych leków przeciwpsychotycznych, praktykowanych przez niektórych narkologów, jest również niedopuszczalne, ponieważ leki te zwiększają prawdopodobieństwo majaczenia, drgawek i działają proarytmicznie. Faktem jest, że po wprowadzeniu do praktyki terapeutycznej fenotiazynowych leków przeciwpsychotycznych liczba zgonów z powodu majaczenia wzrosła kilkukrotnie. Należy unikać jednoczesnego stosowania barbituranów i środków uspokajających, ponieważ połączenie blokerów adrenergicznych i klonidyny może spowodować nadmierną sedację. Powszechnym błędem można nazwać stymulacją w stanach odstawienia wzmożonego oddawania moczu. Z fizjologicznego punktu widzenia taki środek jest nieuzasadniony, gdyż Zespół odstawienia to reakcja układów neuroprzekaźników na zmniejszenie ilości alkoholu we krwi. Zwiększone wydalanie alkoholu prowadzi do ryzyka drgawek i majaczenia. W przypadku ciężkiego odwodnienia lub uporczywych wymiotów wskazana jest płynoterapia. W przypadku niepowikłanych objawów odstawiennych z reguły stosuje się wyłącznie nawadnianie doustne.
W naszym ośrodku stosujemy najskuteczniejsze, legalne i bezpieczne leki oraz metody leczenia. Nasi specjaliści pomogą Ci stworzyć indywidualny algorytm leczenia.
54. Porównaj działanie naloksonu i naltreksonu.
Obie te substancje są konkurencyjnymi antagonistami receptorów opioidowych. Nalokson ma bardzo krótki okres półtrwania, natomiast sen naltrecu trwa kilka dni
Który antagonista receptora opioidowego jest bardziej aktywny niż drugi w łagodzeniu objawów przedawkowania opioidowych leków przeciwbólowych u pacjentów bez uzależnienia od narkotyków?
Nalmefen jest czystym antagonistą opioidów do podawania pozajelitowego.
56. Podaj prosty sposób zdiagnozowania przedawkowania opioidów.
Podawanie naloksonu. Jest krótkotrwały i powinien szybko odwrócić zwężenie źrenic spowodowane przez opioidy, ale nie inne substancje.
57. Dlaczego opioidy stosuje się w celu łagodzenia objawów odstawienia? „amirekson?
Naltrekson jest szczególnie wskazany w przypadku ambulatoryjnego odstawienia opioidów, ponieważ ma długi okres półtrwania (T/= 10 godzin), a jego jednorazowe podanie może blokować euforyczne działanie opioidów na 48 godzin. Zapobiega to próbom nielegalnego pozyskiwania opioidów i wykorzystywania ich do osiągnięcia euforii.
58. Czy nalokson podaje się doustnie?
NIE. Maksymalny efekt występuje po podaniu pozajelitowym, ponieważ nalokson ulega zniszczeniu podczas pierwszego przejścia przez wątrobę.
59. Opisz metabolizm naloksonu.
Nalokson jest metabolizowany głównie przez cytochrom wątrobowy P4^o. Następnie metabolity łączą się z kwasem glukuronowym.
69. Jak podaje się naltrekson?
Naltrekson można podawać doustnie lub pozajelitowo.
61. Opowiedz nam o możliwym zastosowaniu naltreksonu w leczeniu odstawienia alkoholu.
Niedawno wykazano, że naltrekson zmniejsza głód alkoholu i może być stosowany w leczeniu przewlekłego alkoholizmu. Działanie leku jest prawdopodobnie częściowo spowodowane zmianami w aktywności współczulnego układu nerwowego.
62. Opisać skutki naloksonu w ciężkim zatruciu opioidami.
Nalokson szybko przywraca przytomność, jednak ze względu na krótki okres półtrwania pacjenci z ciężkim zatruciem opioidami mogą powrócić do stanu śpiączki w ciągu 60-90 minut. W takim przypadku wskazane jest zastosowanie środka dłużej działającego.
63. Wymień wskazania do stosowania nalmefenu.
Nalmefen, pochodna naltreksonu, jest stosowany w leczeniu przedawkowania opioidów.
^ Jaki jest czas działania nalmefenu?
Substancja tfTO ma długi okres półtrwania (8-10 godzin) i działa dość długo.
65- Czy działanie antagonistów receptorów opioidowych występuje na ulicach, na których nie stosuje się opioidów?
Neji: Substancje te są praktycznie obojętne, co pozwala na ich zastosowanie w diagnostyce przedawkowania opioidów bez obawy o możliwość wystąpienia skutków ogólnoustrojowych.
66. Opisać dynamikę zanikania działania opioidów po dożylnym podaniu antagonisty opioidów.
Zanik działania opioidów obserwuje się w ciągu 1-3 minut. Rozmiary źrenic, oddychanie i świadomość przywracane są bardzo szybko.
67. Czy antagoniści receptorów opioidowych uzależniają?
NIE. Przy długotrwałym stosowaniu uzależnienie nie rozwija się, a odstawieniu tych ulicznych substancji bez uzależnienia od opioidów nie towarzyszą żadne negatywne objawy.
68. Czy antagoniści mają taką samą aktywność na wszystkich receptorach opioidowych?
NIE. Substancje te z reguły najaktywniej oddziałują z receptorami mu i w różnym stopniu wpływają na inne typy receptorów
FARMAKOLOGIA SERCA NACZYNIOWEGO
SYSTEMY Patricia K. Anthony, Judith Kautz,
Andrew Powersowi i Rebece Tome
Problem otyłości w połączeniu z różnymi zaburzeniami metabolicznymi jest przedmiotem zainteresowania współczesnej medycyny i opieki zdrowotnej, gdyż prowadzi do rozwoju szeregu poważnych chorób obniżających jakość życia i zwiększających śmiertelność wśród ludności pracującej. Zatem ryzyko cukrzycy typu 2 (DM) wzrasta 2-krotnie w przypadku otyłości klasy I, 5-krotnie w przypadku otyłości klasy II i ponad 10-krotnie w przypadku otyłości klasy III–IV. Ponadto powszechnie wiadomo, że u ponad 80% chorych na cukrzycę typu 2 występuje otyłość o różnym stopniu nasilenia. Nadwaga i otyłość są również czynnikami ryzyka choroby niedokrwiennej serca. W badaniu prospektywnym Ischemic Heart Disease Risk Factors Study wykazano, że wśród pacjentów z zespołem metabolicznym choroba niedokrwienna serca rozwijała się 3–4 razy częściej, a śmiertelność z powodu tej choroby była 3–5 razy większa w porównaniu z pacjentami bez zaburzeń metabolicznych. Niebezpieczeństwo otyłości wiąże się ze zwiększonym ryzykiem wystąpienia nadciśnienia tętniczego (AH), udaru niedokrwiennego mózgu, zespołu bezdechu sennego, nowotworów złośliwych niektórych lokalizacji (rak okrężnicy, piersi, endometrium itp.) oraz choroby zwyrodnieniowej stawów, a także ma negatywny wpływ na zdrowie psychospołeczne pacjentów i jakość ich życia. Według ekspertów Światowej Organizacji Zdrowia (WHO) w samej Europie z powodu chorób związanych z otyłością umiera rocznie 320 tys. osób. Wykazano, że wyższy poziom otyłości wiąże się ze zwiększoną śmiertelnością, przede wszystkim z powodu chorób układu krążenia, cukrzycy i niektórych rodzajów nowotworów.
W leczeniu otyłości podstawowe i patogenetycznie uzasadnione działania mają na celu normalizację zaburzeń metabolicznych i redukcję masy ciała. Obecnie skuteczność leczenia pacjentów otyłych pozostaje niezwykle niska, gdyż u większości pacjentów masa ciała zmniejsza się bardzo powoli, przyjmują oni postawę pasywną na etapie stabilizacji zredukowanej masy ciała. Nie można nie zauważyć pesymizmu lekarzy dotyczącego wysiłków pacjentów zmierzających do utraty wagi. Należy szczególnie podkreślić, że celowa utrata masy ciała, powiązana z obniżeniem ciśnienia krwi (BP), u pacjentów z nadciśnieniem tętniczym, prowadzi do poprawy profilu lipidowego i zmniejszenia zachorowalności na cukrzycę. W artykule przeglądowym przedstawiono problematykę farmakoterapii otyłości z perspektywy historycznej.
Leki zmniejszające apetyt lub zwiększające uczucie sytości działają na różne neuroprzekaźniki ośrodkowego układu nerwowego (OUN) (noradrenergiczne i serotoninergiczne).
Sympatykomimetyki tłumią apetyt poprzez stymulację uwalniania noradrenaliny i dopaminy z zakończeń nerwowych w ośrodku nasycenia podwzgórza. Inne skutki wywoływane przez sympatykomimetyki, takie jak zahamowanie wydzielania soku żołądkowego i zwiększenie wydatku energetycznego, mogą również przyczyniać się do zmniejszenia apetytu i utraty masy ciała. Leki działające ośrodkowo, takie jak fentermina, dietylopropion, fendimetrazyna, benzfetamina i mazindol, są zatwierdzone do leczenia otyłości w Stanach Zjednoczonych. Co więcej, fentermina jest najczęściej przepisywanym lekiem. Jednocześnie leki te nie znajdują się na liście leków z wyboru w leczeniu otyłości. We współczesnych schematach leczenia przepisywanie sympatykomimetyków ogranicza się do kilku tygodni ze względu na ryzyko wystąpienia uzależnienia od narkotyków, chociaż istnieją dane dotyczące dłuższego ich stosowania (6 miesięcy i dłużej). Skutki uboczne tej grupy leków obejmują bezsenność, suchość w ustach, zaparcia, euforię, kołatanie serca i podwyższone ciśnienie krwi. Leki noradrenergiczne są przeciwwskazane w przypadku ciężkiej miażdżycy, chorób naczyń mózgowych, umiarkowanego i ciężkiego nadciśnienia, tyreotoksykozy, jaskry, pobudzenia psychicznego lub uzależnienia od narkotyków w wywiadzie.
Fentermina należy do rodziny b-fenyloetyloamin; zatwierdzony w 1959 roku przez Agencję ds. Żywności i Leków (FDA) do krótkotrwałego (do 3 miesięcy) stosowania w leczeniu otyłości. Nie ma wystarczających danych dotyczących skuteczności i bezpieczeństwa fenterminy podczas długotrwałego stosowania, zwłaszcza w monoterapii. Obecnie w rutynowej praktyce klinicznej w leczeniu otyłości stosowanie leków z rodziny b-fenyloetyloamin jest ograniczone i nie są stosowane w terapii długotrwałej. W kontrolowanym badaniu klinicznym leczenie fenterminą przez 36 tygodni spowodowało zmniejszenie masy ciała o 12,2 kg w porównaniu z 4,8 kg w grupie placebo (p.< 0,001). По данным метаанализа, включавшего 6 рандомизированных клинических исследований длительностью от 2 до 24 нед, на фоне терапии фентермином наблюдалось дополнительное снижение массы тела в среднем на 3,6 кг по сравнению с плацебо. В двойном слепом плацебо-контролируемом исследовании 74 пациента с контролируемым СД, АГ или дислипидемией, страдающих ожирением, были рандомизированы на прием фентермина с контролируемым высвобождением (в дозе 30 мг/сут) или плацебо. Через 12 нед лечения фентермином показано существенное уменьшение массы тела (на 9,3 ± 3,4 кг против 1,8 ± 3,1 кг, р < 0,001) и окружности талии (7,2 ± 0,5 см против 2,1 ± 0,6 см, р < 0,001) по сравнению с группой плацебо. В группе фентермина с контролируемым высвобождением клинически значимого снижения массы тела (≥ 5%) достигли 95,8% пациентов (против 20,8% в группе плацебо, p < 0,001), 62,5% уменьшили массу тела более чем на 10% от исходной (против 4,7% в группе плацебо, р < 0,001). Продемонстрировано благоприятное влияние фентермина с контролируемым высвобождением на содержание общего холестерина (ХС) и ХС липопротеинов низкой плотности (ЛПНП). Существенных различий по систолическому и диастолическому АД между группами не отмечено, тогда как частота сердечных сокращений (ЧСС) значительно увеличивалась в основной группе по сравнению с плацебо (р = 0,02).
Najczęstszymi działaniami niepożądanymi były suchość w ustach i bezsenność, które były przemijające. Podsumowując, krótkotrwałe leczenie fenterminą o kontrolowanym uwalnianiu spowodowało znaczną redukcję masy ciała i obwodu talii oraz poprawę parametrów profilu lipidowego, a także brak poważnych skutków ubocznych. Ze względu na fakt, że fentermina jest sympatykomimetykiem, należy liczyć się z możliwością wystąpienia działań niepożądanych, takich jak bezsenność, suchość w ustach, zawroty głowy, kołatanie serca, drżenie rąk, podwyższone ciśnienie krwi i częstość akcji serca. Dlatego podczas przepisywania leków sympatykomimetycznych zaleca się monitorowanie ciśnienia krwi i tętna.
Dietylopropion podobna do amfetaminy, ale różni się od tej ostatniej nieznacznie wyrażoną aktywnością sympatykomimetyczną i mniejszą liczbą skutków ubocznych. W Stanach Zjednoczonych dietylopropion został zatwierdzony do leczenia otyłości w 1959 r. Metaanaliza 13 badań oceniających skuteczność terapii dietylopropionem u otyłych pacjentów przez średnio 20 tygodni wykazała dodatkową utratę masy ciała o 3,0 kg w porównaniu z placebo. W brazylijskim badaniu oceniano skuteczność i tolerancję dietylopropionu przez 1 rok leczenia. Po 2 tygodniach badań przesiewowych 69 otyłym pacjentom (wskaźnik masy ciała (BMI) 30–45 kg/m2) przepisano dietę niskokaloryczną, a następnie losowo przydzielono do grupy otrzymującej dietylopropion w dawce 100 mg/dobę (37 pacjentów) lub placebo (32 pacjentów). przez 6 miesięcy Następnie badanie stało się badaniem otwartym i wszystkim pacjentom przepisano dietylopropion na kolejne 6 miesięcy. Po pierwszych 6 miesiącach zaobserwowano istotny spadek masy ciała w grupie dietylopropionu o 9,8% (średnio 9,3 kg), natomiast w grupie placebo spadek wyniósł 3,7% (3,1 kg), co było statystycznie istotne . Po 12 miesiącach w grupie początkowo przyjmującej lek spadek masy ciała wyniósł 10,6% (10,1 kg), natomiast w grupie, która po 6 miesiącach przeszła na dietylopropion – 7,0% (średnio 6,7 kg). Analiza ciśnienia krwi, tętna, badania elektrokardiograficzne i psychologiczne nie wykazały istotnych różnic pomiędzy grupami. W grupie dietylopropionu istotnie częściej obserwowano jedynie takie skutki uboczne jak suchość w ustach i bezsenność i to tylko w ciągu pierwszych 3 miesięcy.
Fentermina i dietylopropion zaliczane są do IV klasy (według klasyfikacji amerykańskiej Agencji Leków), co wskazuje na niskie ryzyko nadużywania tych leków i zapewnia największe bezpieczeństwo dla pacjentów.
Leki serotoninergiczne(fenfluramina, deksfenfluramina) zwiększają stężenie serotoniny w mózgu poprzez hamowanie jej wychwytu zwrotnego. Znaczący spadek masy ciała w ciągu 1 roku, z największym efektem w ciągu pierwszych 6 miesięcy, wynika ze zmniejszenia dziennego spożycia energii o 10–15%. W 1997 roku oba te leki zostały wycofane z rynku leków ze względu na rozwój patologii zastawek serca i nadciśnienia płucnego.
Sibutraminałączy w sobie działanie inhibitora wychwytu zwrotnego serotoniny, noradrenaliny i dopaminy. Początkowo lek przeszedł badania kliniczne jako lek przeciwdepresyjny, podczas których ujawniono jego wyraźne działanie anoreksogenne. Sibutramina i jej aktywne metabolity hamują wychwyt zwrotny serotoniny i noradrenaliny, przedłużając w ten sposób interakcję tych neuroprzekaźników z ich receptorami postsynaptycznymi. W rezultacie zwiększa się i przedłuża uczucie sytości, co powoduje zmniejszenie ilości spożywanego pokarmu, a co za tym idzie, zmniejszenie spożycia energii. Jednocześnie lek jest słabym inhibitorem wychwytu zwrotnego dopaminy. W przeciwieństwie do fenfluraminy i deksfenfluraminy, sibutramina nie wzmaga uwalniania serotoniny i nie powoduje zaburzeń zastawek. Dodatkowo sibutramina zwiększa wydatek energetyczny na skutek wzmożonej termogenezy, co zwiększa zdolność leku do redukcji masy ciała.
W leczeniu otyłości sibutramina została dopuszczona do użytku medycznego w Meksyku w 1997 r., po czym została zarejestrowana w 80 krajach. Metaanaliza szeregu randomizowanych, kontrolowanych placebo badań skuteczności sibutraminy, które obejmowały pacjentów otyłych przez 12 miesięcy, wykazała spadek masy ciała przewyższający grupę placebo o 4,2–4,45 kg. Jak wynika z licznych badań, podczas terapii sibutraminą w dawce 10–15 mg/dobę przez 12 miesięcy zaobserwowano skuteczny i klinicznie istotny spadek masy ciała (o 5–10%) u ponad 86% pacjentów z różnymi stopnie otyłości. Dodanie sibutraminy do standardowej terapii nielekowej spowodowało istotnie większą redukcję masy ciała (o 11,3 kg w ciągu pierwszych 6 miesięcy leczenia) niż sama modyfikacja stylu życia. Jednocześnie trzewna tkanka tłuszczowa uległa korzystnemu zmniejszeniu. Podczas terapii sibutraminą spektrum lipidów osocza krwi uległo poprawie wraz ze zmianą kierunku w kierunku przeciwmiażdżycowym (obniżenie poziomu trójglicerydów i zwiększenie stężenia cholesterolu lipoprotein o dużej gęstości (HDL)), a także zmniejszenie stężenia glukozy w osoczu krwi na czczo i poziomu insuliny . Jednocześnie w metaanalizie obejmującej 10 badań, w których łącznie wzięło udział 1213 uczestników przyjmujących sybutraminę lub placebo przez 6–12 miesięcy, nie stwierdzono związku pomiędzy leczeniem sibutraminą a zmniejszeniem poziomu cholesterolu całkowitego po uwzględnieniu utraty masy ciała. . Ogólnie rzecz biorąc, sibutramina była dobrze tolerowana przez pacjentów. Działania niepożądane obserwowane w przypadku sibutraminy obejmują suchość w ustach, ból głowy, bezsenność i zaparcia. Najbardziej znaczącymi działaniami niepożądanymi leku było zwiększenie ciśnienia krwi i częstości akcji serca. Tym samym w trakcie leczenia sibutraminą zaobserwowano spadek skuteczności terapii hipotensyjnej.
W początkowej fazie powszechnego stosowania sibutramina była powodem wielu dyskusji i decyzji administracyjnych w wielu krajach w związku z podejrzeniami rozwoju poważnych powikłań, głównie ze strony układu sercowo-naczyniowego. W 2002 roku rozpoczęło się badanie SCOUT (Sibutraamine Cardionaczyniowe Outcome Trial), w którym wzięło udział 10 742 pacjentów w 300 ośrodkach medycznych zlokalizowanych w 16 krajach. Celem badania była ocena stosunku skuteczności do bezpieczeństwa stosowania sibutraminy u osób z otyłością wysokiego ryzyka (97% miało choroby układu krążenia, 88% nadciśnienie, 84% cukrzycę typu 2). Wykazano, że u osób z chorobami układu krążenia długotrwałe (5 lat) leczenie sybutraminą przyczyniło się do istotnego wzrostu ryzyka zawału mięśnia sercowego niezakończonego zgonem (o 16%) oraz udaru mózgu niezakończonego zgonem. Jednakże średnia różnica pomiędzy masą ciała pacjentów otrzymujących sibutraminę i placebo wynosiła jedynie 2,5%. Uznano, że ten stosunek korzyści do ryzyka jest niedopuszczalny i Europejska Agencja Leków (EMEA) zaleciła zawieszenie wprowadzenia sybutraminy do obrotu w Unii Europejskiej. W październiku 2010 r. firma Abbott Laboratories wycofała oryginalny lek sibutraminę z rynków Stanów Zjednoczonych i Unii Europejskiej ze względu na zwiększone ryzyko zawału mięśnia sercowego i udaru mózgu.
Inne leki o działaniu anoreksogennym
Antagoniści receptora kannabinoidowego CB 1
Obecnie wiele uwagi poświęca się układowi endokannabinoidowemu, który zajmuje kluczowe miejsce w patogenezie chorobliwej otyłości. Wykazano ścisły związek pomiędzy receptorami układu endokannabinoidowego a substancjami biologicznie czynnymi trzewnej tkanki tłuszczowej. Udowodniono regulacyjną rolę układu endokannabinoidowego w kontroli apetytu oraz metabolizmie glukozy i lipidów.
Rymonabant- pierwszy przedstawiciel nowej klasy leków - blokery receptorów kannabinoidowych pierwszego typu (CB 1). Poprzez selektywne wiązanie się z centralnymi i obwodowymi receptorami CB1, rimonabant moduluje nadpobudliwy układ endokannabinoidowy. Wyniki programu RIO, który obejmował 4 badania III fazy kontrolowane placebo metodą podwójnie ślepej próby, w których wzięło udział ponad 6 tys. pacjentów z nadwagą lub otyłością, były podobne: przyjmowanie rimonabantu przez 1-2 lata doprowadziło do istotnego statystycznie spadku masy ciała, a także znaczną mobilizację brzusznej tkanki tłuszczowej, która objawiała się dość wyraźnym zmniejszeniem obwodu talii. Ponadto u pacjentów z nadwagą lub otyłością, z cukrzycą typu 2 i bez niej, stwierdzono pozytywny wpływ rimonabantu na kardiometaboliczne czynniki ryzyka, w szczególności na stężenie triglicerydów, cholesterolu HDL, białka C-reaktywnego, ciśnienie krwi, insulinooporność, a także dobra tolerancja leku. Jednak nowsze doniesienia wskazują, że stosowanie rimonabantu wiąże się ze zwiększonym ryzykiem wystąpienia zaburzeń psychicznych, w tym lęku, depresji i myśli samobójczych. I tak, według czterech badań, niekorzystne zdarzenia psychiczne odnotowano u 26% uczestników grupy rimonabant w porównaniu do 14% pacjentów w grupie placebo, a ryzyko wystąpienia zaburzeń depresyjnych było 2,5 razy większe niż w grupie placebo. Zdaniem ekspertów pojawienie się objawów zaburzeń psychicznych podczas stosowania modyfikatorów (antagonistów lub odwracalnych agonistów) receptorów kannabinoidowych jest całkiem spodziewane z ogólnego biologicznego punktu widzenia, ponieważ endokannabinoidy są ważnymi modulatorami w stanach patologicznych, takich jak stany lękowe, depresja, zaburzenia stresowe pourazowe i fobie. Wyższy był również wskaźnik samobójstw, w tym myśli samobójczych: iloraz szans = 2,0 (1,2 do 3,4) przy różnicy ryzyka 0,34 (0,14 do 0,54) w porównaniu z placebo. Jednocześnie pojawiają się zaburzenia neurologiczne i żołądkowo-jelitowe w postaci zawrotów głowy, nudności i biegunki. Ostatecznie w czerwcu 2007 roku producent komercyjnego produktu farmaceutycznego rimonabant wycofał swój wniosek o dopuszczenie do obrotu w USA w związku z zaleceniem FDA, aby nie dopuścić go do sprzedaży w kraju ze względu na konieczność dalszych badań nad skutkami ubocznymi, a w listopadzie W 2008 r. EMEA wycofała zgodę na stosowanie rymonabantu w Europie.
Leki przeciwdepresyjne
Istnieją dwa leki zmniejszające masę ciała, które można stosować w leczeniu otyłości jedynie w przypadku specjalnych wskazań. Jeden z nich jest fluoksetyna- selektywny inhibitor wychwytu zwrotnego serotoniny, selektywnie blokuje wychwyt zwrotny serotoniny (5-HT) w synapsach neuronów OUN, zmniejsza apetyt, co może prowadzić do utraty wagi. W krótkich badaniach obserwacyjnych kontrolowanych placebo zmniejszał masę ciała w dawce 20–40 mg/dzień (średnio 5%). W badaniach z zastosowaniem fluoksetyny w dawce 60 mg/dobę przez 6–8 tygodni zgłaszano istotne zmniejszenie masy ciała, przy czym maksymalny efekt osiągano po 12–20 tygodniach, a następnie zwiększano. Przy długotrwałym stosowaniu leku (52 tygodnie) nie stwierdzono istotnej różnicy w działaniu na masę ciała pomiędzy grupą fluoksetyny i placebo. Jednocześnie 8-miesięczna terapia skojarzona fluoksetyną i deksfenfluraminą spowodowała istotnie większą utratę masy ciała w porównaniu z placebo (13,4 wobec 6,2 kg w grupie placebo). Istnieją doniesienia o stosowaniu w praktyce klinicznej kombinacji fluoksetyny i fenterminy, jednak brak jest dowodów na jej skuteczność i bezpieczeństwo w długotrwałym leczeniu. Wskazaniami do stosowania fluoksetyny są bulimia psychiczna, depresja dietetyczna oraz obecność zaburzeń depresyjnych lub lękowo-depresyjnych u osób otyłych. Skutki uboczne fluoksetyny obejmują bóle głowy, osłabienie, nudności, biegunkę, senność, bezsenność, nerwowość, pocenie się i drżenie.
Kolejnym lekiem jest lek przeciwdepresyjny bupropion, zmniejszając uzależnienie od nikotyny u palaczy. Jego głównym działaniem farmakologicznym jest selektywne hamowanie wychwytu zwrotnego noradrenaliny i dopaminy. Jest wybiórczo wychwytywana przez transporter dopaminy (DAT), jednak główny efekt terapeutyczny wynika z hamowania wychwytu zwrotnego noradrenaliny. Działa również jako antagonista nikotynowych receptorów acetylocholiny. To skłoniło do przeprowadzenia badania klinicznego mającego na celu ocenę potencjału bupropionu w postaci preparatu o powolnym uwalnianiu w leczeniu otyłości.
Bupropion, pierwotnie opracowany i sprzedawany jako lek przeciwdepresyjny, wkrótce okazał się skuteczny w leczeniu uzależnienia od nikotyny. W szeregu badań wykazano, że bupropion w dawce 100–300 mg/dobę powoduje nieznaczny spadek masy ciała (około 5%). Metaanaliza leków przeciw otyłości, obejmująca trzy badania bupropionu z placebo i podwójnie ślepą próbą, wykazała skuteczność bupropionu w dawce 400 mg/dobę w leczeniu otyłości. Zatem w okresie 6–12 miesięcy średni spadek masy ciała w grupie przyjmującej bupropion był istotnie większy (4,4 kg) niż w grupie placebo (1,7 kg). Dodatkowo, statystycznie identyczne wyniki utraty wagi zaobserwowano podczas przyjmowania bupropionu i innych leków jego zmniejszających, takich jak sibutramina, orlistat i dietylopropion. Jednak pomimo wyraźnego spadku masy ciała, stosowanie bupropionu spowodowało wzrost częstości występowania nadciśnienia tętniczego. Wskazaniami do przyjmowania bupropionu są depresja spowodowana otyłością oraz sytuacja, gdy długotrwale otyły pacjent palący ma zamiar rzucić palenie.
Poniżej przedstawiono ocenę skuteczności połączenia bupropionu z zonisamidem i bupropionu z naltreksonem.
Leki zmniejszające wchłanianie składników odżywczych
Orlistat- pierwszy i jak dotąd jedyny lek działający obwodowo, stosowany w praktyce klinicznej w leczeniu otyłości od 1998 roku. Orlistat jest syntetyczną pochodną lipstatyny, produktu odpadowego grzyba pleśniowego Streptomyces toksyczne, który hamuje lipazy żołądkowe i trzustkowe. Lipazy żołądkowo-jelitowe to kluczowe enzymy biorące udział w hydrolizie trójglicerydów z pożywienia, uwalniając kwasy tłuszczowe i monoglicerydy, które następnie są wchłaniane przez błonę śluzową jelit. Ze względu na strukturalne podobieństwo orlistatu do trójglicerydów, lek oddziałuje z miejscem aktywnym enzymu, wiążąc się kowalencyjnie z jego resztą seryny. Wiązanie jest powoli odwracalne, jednak w warunkach fizjologicznych działanie hamujące leku podczas przejścia przez przewód pokarmowy (GIT) pozostaje niezmienione. W rezultacie około jedna trzecia trójglicerydów znajdujących się w pożywieniu nie jest trawiona ani wchłaniana, co pozwala na wytworzenie dodatkowego deficytu kalorycznego w porównaniu do samej diety. Orlistat nie wpływa jednak na metabolizm węglowodanów, białek i fosfolipidów. Zatem orlistat ma lokalny mechanizm działania ograniczony do przewodu żołądkowo-jelitowego. Mniej niż 1% orlistatu przedostającego się do przewodu pokarmowego ulega wchłonięciu, zatem nie ma on ogólnoustrojowego działania na lipazy. Około 97% podanej dawki leku było wydalane z kałem, z czego 87% stanowił orlistat w postaci niezmienionej. Skuteczność orlistatu jest optymalna, jeśli jest przyjmowany w trakcie lub w ciągu 1 godziny po posiłku zawierającym mniej niż 30% kalorii pochodzących z tłuszczu. Orlistat przyjmuje się 3 razy dziennie po 120 mg podczas posiłku lub w ciągu 1 godziny po jego przyjęciu.
Skuteczność orlistatu w redukcji masy ciała wykazano w szeregu randomizowanych badaniach klinicznych. Badanie przeprowadzone przez Sjostroma i wsp. z udziałem 743 otyłych pacjentów, sprawdzające skuteczność orlistatu, wykazało utratę masy ciała, a następnie jej utrzymanie. Badania wykazały, że przy stosowaniu orlistatu następuje nie tylko ogólne zmniejszenie masy tkanki tłuszczowej, ale także zmniejszenie masy tkanki tłuszczowej trzewno-brzusznej. Pomaga to zwiększyć wrażliwość na insulinę i zmniejszyć hiperinsulinemię, co stanowi skuteczną profilaktykę rozwoju cukrzycy typu 2. W 4-letnim, podwójnie ślepym, kontrolowanym placebo badaniu XEDOS (XENical in the Prevention of Diabetes in Obese Objects), w którym wzięło udział 3305 pacjentów otyłych (BMI ≥ 30 kg/m2) z prawidłową (79%) lub upośledzoną (21) %) tolerancji glukozy, badano skuteczność orlistatu w połączeniu z modyfikacją stylu życia w profilaktyce cukrzycy typu 2. Wykazano, że połączenie orlistatu z modyfikacją stylu życia spowodowało zmniejszenie masy ciała o 5,8 kg w porównaniu z 3,0 kg w grupie placebo oraz istotne zmniejszenie ryzyka zachorowania na cukrzycę typu 2 (6,2 w porównaniu z 9% w grupie placebo). Jednocześnie skumulowana zapadalność na cukrzycę typu 2 w grupie głównej była o 37,3% niższa niż w grupie kontrolnej. W kilku badaniach prowadzonych metodą podwójnie ślepej próby, kontrolowanych placebo, czas stosowania orlistatu wynosił 2 lata. Po 12 miesiącach leczenia odnotowano istotny spadek masy ciała wynoszący 2,89 kg (skorygowany o zmiany masy ciała w grupie kontrolnej). Największy spadek masy ciała zaobserwowano w ciągu pierwszych 6 miesięcy leczenia, a następnie w trakcie dalszego podawania leku utrzymywał się on na stałym poziomie i mniejszym niż w grupie kontrolnej.
Diety bardzo niskoenergetyczne (VLED; 400–800 kcal/dzień) zawierające znaczne ilości białka mogą powodować znaczną utratę wagi w krótkim okresie, ale dane dotyczące długoterminowego utrzymania są na ogół rozczarowujące. W tym kontekście bardzo ważne jest, jak długo orlistat zapobiega przyrostowi masy ciała po stosowaniu leku ONED u pacjentów z otyłością i metabolicznymi czynnikami ryzyka. W badaniu klinicznym pacjentów, u których pierwotną utratę masy ciała uzyskano dzięki stosowaniu preparatu ONED, losowo przydzielono do grupy otrzymującej orlistat lub placebo przez 3 lata. Spadek masy ciała po 8 tygodniach stosowania ONED wyniósł 14,3 ± 2,0 kg w grupie orlistatu i 14,5 ± 2,1 kg w grupie placebo. Przyrost masy ciała w ciągu 36 miesięcy po zakończeniu ONED był istotnie mniejszy w grupie orlistatu (4,6 ± 8,6 vs. 7,0 ± 7,1 kg; p< 0,02). Поддержание массы тела сопровождалось существенным улучшением ряда метаболических параметров. Так, ретроспективный анализ показал, что лечение орлистатом приводило к снижению уровня триглицеридов и общего ХС в плазме крови, улучшению толерантности к глюкозе, снижению систолического и диастолического АД.
Leczenie otyłości u dzieci i młodzieży jest zadaniem złożonym; zmiany stylu życia w wielu przypadkach nie prowadzą do istotnej klinicznie redukcji masy ciała, zwłaszcza u młodzieży. Według ekspertów bez wsparcia farmakologicznego nie więcej niż 4–5% dzieci może osiągnąć znaczną utratę masy ciała. Dlatego w ostatnie lata Badania były i są prowadzone nad skutecznością i bezpieczeństwem stosowania leków w tej grupie wiekowej. W kilku badaniach oceniano skuteczność orlistatu u młodzieży. W badaniu z podwójnie ślepą próbą, kontrolowanym placebo, z udziałem 539 otyłych nastolatków w wieku 12–16 lat, po 1 roku leczenia BMI zmniejszyło się o 0,55 kg/m2 w grupie orlistatu i wzrosło o 0,31 kg/m2 w grupie placebo ( p = 0,001). Zmiana obwodu talii była następująca: spadek w grupie głównej i wzrost w grupie placebo. Jednakże w innym podwójnie ślepym, randomizowanym badaniu kontrolowanym placebo z udziałem 40 nastolatków, 6-miesięczne leczenie orlistatem nie miało istotnego wpływu na BMI. Dlatego potrzebne są dalsze badania w tym kierunku.
Działania niepożądane orlistatu ograniczają się do objawów żołądkowo-jelitowych i występują u około 15–30% pacjentów. Do działań niepożądanych orlistatu zalicza się tłustą wydzielinę z odbytu, tłuste stolce, wzmożone wypróżnienia, potrzebę wypróżnienia i wzdęcia. Zazwyczaj zjawiska te mają charakter łagodny lub umiarkowany, ich częstotliwość maleje w miarę wydłużania się czasu leczenia, jednak w prawie 9% przypadków stają się przyczyną zaprzestania stosowania orlistatu. 7% pacjentów otrzymujących orlistat zgłosiło objawy nietrzymania stolca w porównaniu z 1% w grupie placebo. Stosowanie orlistatu może upośledzać wchłanianie witamin rozpuszczalnych w tłuszczach (A, D, E i K) oraz b-karotenu, dlatego zaleca się profilaktyczną suplementację witamin. Ogólnoustrojowe działania niepożądane orlistatu występują niezwykle rzadko ze względu na brak wchłaniania ogólnoustrojowego.
Zwiększone spożycie tłuszczów w okrężnicy wzbudziło obawy dotyczące zwiększonego ryzyka raka okrężnicy. Konieczne są dalsze badania w tym kierunku. Ponadto inhibitory lipazy mogą zwiększać wchłanianie szczawianów i zwiększać ryzyko kamicy nerkowej i niewydolności nerek.
Perspektywy farmakoterapii otyłości
Pramlintyd- syntetyczny analog hormonu trzustki amyliny, pierwotnie syntetyzowany jako lek stosowany w leczeniu cukrzycy typu 1 i typu 2. W USA lek jest dopuszczony do stosowania jako terapia wspomagająca insulinę. Pramlintyd podaje się podskórnie przed posiłkami. Lek hamuje zależne od glukozy wytwarzanie glukagonu i przede wszystkim zmniejsza poposiłkowe wahania glikemii. Następnie ustalono związek ze zmniejszonym apetytem, przyjmowaniem pokarmu i szybką sytością związaną z motoryką przewodu pokarmowego. Obecnie jest badany jako potencjalny lek do leczenia otyłości. 16-tygodniowe randomizowane badanie kliniczne ze zwiększaniem dawki wykazało znacznie większą redukcję masy ciała w grupie otrzymującej pramlintyd w dawce 240 mcg o 3,7% w porównaniu z placebo (p< 0,001); доля пациентов с уменьшением массы тела ≥ 5% составила 31% пациентов (p < 0,001) . В клиническом исследовании , включавшем 411 пациентов с ожирением, рандомизированных на прием прамлинтида (6 подгрупп в дозах 120, 240 и 360 мкг 2 и 3 раза в сутки) или плацебо в течение 4 мес, и далее продленном до 1 года. Уменьшение массы тела восстанавливалось в группе плацебо, но сохранялось во всех группах прамлинтида, кроме лиц, получавших препарат в дозе 120 мг 2 раза в сутки. Наиболее частым побочным эффектом была тошнота.
Analogi peptydów glukagonopodobnych
Obecnie nowe podejścia terapeutyczne w leczeniu otyłości wiążą się z modulacją aktywności poziomu peptydu glukagonopodobnego (GLP-1) poprzez podawanie analogów i mimetyków GLP-1 (eksenatyd, liraglutyd, CJC-1131), opracowany i zatwierdzony do leczenia cukrzycy typu 2. Ta klasa leków charakteryzuje się podwójnym mechanizmem działania, a mianowicie wpływem na przewód pokarmowy i mózg. Tym samym z przewodu pokarmowego do mózgu wysyłane są sygnały stymulujące wydzielanie leptyny, kluczowego mediatora pomiędzy tkanką tłuszczową a układem podwzgórzowo-przysadkowym, co prowadzi do zmniejszenia apetytu, zużycia energii i szybkości opróżniania żołądka. Badania na modelach zwierzęcych i zdrowych ochotnikach wykazały, że GLP-1 jest jednym z ważnych regulatorów ilości spożywanego pokarmu, zwiększa uczucie sytości i zmniejsza uczucie głodu. Główną korzyścią z długotrwałego stosowania liraglutydu i eksenatydu jest zmniejszenie stężenia hemoglobiny glikowanej (HbA1C) i skurczowego ciśnienia krwi.
Liraglutyd- analog ludzkiego GLP-1, wytwarzany w drodze biotechnologii rekombinowanego kwasu dezoksyrybonukleinowego przy użyciu szczepu Saccharomyces cerevisiae, który wykazuje 97% homologii z ludzkim GLP-1, który wiąże i aktywuje receptory GLP-1 u człowieka. Receptor GLP-1 służy jako cel dla natywnego GLP-1, endogennego hormonu inkretyny, który powoduje stymulację zależnego od glukozy wydzielania insuliny w komórkach b trzustki. W badaniu prowadzonym metodą podwójnie ślepej próby, kontrolowanym placebo, z udziałem 564 otyłych pacjentów z wysokim ryzykiem rozwoju cukrzycy, porównywano liraglutyd z inhibitorem lipazy, orlistatem. Czas trwania badania wynosił 20 tygodni. Ustalono, że dzienne spożycie liraglutydu w dawce 1,2; 1,8; dawki 2,4 i 3,0 mg powodowały średnią redukcję masy ciała o 4–8 kg (p = 0,003), 5,5; 6,3 i 7,2 kg (str< 0,0001 для дозирований 1,8–3,0 мг) соответственно. При этом в группе плацебо уменьшение массы тела составило 2,8 кг, а в группе орлистата - 4,1 кг. Лечение лираглутидом в наиболее высокой дозе приводило к уменьшению массы тела на ≥ 5% у 75% пациентов и на ≥ 10% - у более 25% обследованных больных. Кроме того, выявлено благоприятное действие лираглутида на уровень ХС ЛПНП в плазме крови и систолическое АД. В целом отмечена хорошая переносимость лираглутида, в то же время 10% пациентов были исключены из исследования вследствие развития побочных действий. Побочные эффекты лираглутида отмечались в основном со стороны ЖКТ, и большинство из них были оценены как умеренные. Наиболее частыми нежелательными явлениями в группе лираглутида были тошнота и рвота. Индивидуальная непереносимость отмечена у 20–50% пациентов, зависела от дозы препарата и проявлялась тошнотой. Ощущение подташнивания при приеме препарата наиболее часто отмечали в самом начале применения.
Eksenatyd to syntetyczny peptyd, którego sekwencja aminokwasów jest w 53% identyczna z sekwencją ludzkiego hormonu, inkretyny GLP-1, dzięki której eksenatyd działa jako silny agonista receptorów GLP-1 u ludzi. Stosowanie eksenatydu u pacjentów z cukrzycą typu 2 i nadwagą lub otyłością prowadzi również do postępującego i trwałego spadku masy ciała. We wszystkich badaniach klinicznych stosowanie eksenatydu powodowało istotne postępujące zmniejszenie masy ciała pacjentów, co u większości pacjentów z nadwagą obserwowano po 2–4 tygodniach leczenia. Zaobserwowany efekt utrzymywał się przez 2 lata leczenia w badaniach otwartych, które były kontynuacją badań III fazy kontrolowanych placebo. Ustalono zależny od dawki wpływ eksenatydu na masę ciała. U osób, które całkowicie ukończyły 2-letni okres badania, terapia eksenatydem w dawce 10 mcg 2 razy dziennie pozwoliła uzyskać redukcję masy ciała o 1,6; 2,4 i 4,7 kg po 12; Odpowiednio 30 i 104 tygodnie leczenia. Utratę masy ciała zaobserwowano u 81% chorych na cukrzycę typu 2, którzy otrzymywali eksenatyd przez 2 lata, mimo że protokół badania nie wymagał szczególnej diety ani programu ćwiczeń.
Zgodnie z wynikami przeglądu systematycznego i metaanalizy, u pacjentów otrzymujących agonistów receptora GLP-1 wykazano istotniejszy spadek masy ciała, a także normalizację ciśnienia krwi i poziomu cholesterolu, niezależnie od obecności cukrzycy typu 2. Metaanaliza objęła 25 randomizowanych, kontrolowanych badań (w sumie 10 560 uczestników), w których pacjenci otrzymywali agonistów GLP-1 (liraglutyd lub eksenatyd) przez co najmniej 20 tygodni. Spadek masy ciała zaobserwowano zarówno u pacjentów bez cukrzycy (średnia ważona różnica -3,2 kg; 95% CI -4,3 do -2,1), jak i chorych na cukrzycę (średnia ważona różnica -2,8 kg; 95% CI -3,4 do -2,3). Co więcej, największy spadek masy ciała wiązał się ze stosowaniem wyższych dawek agonistów GLP-1. Analiza podgrup pacjentów otrzymujących eksenatyd dwa razy na dobę (-2,8 kg; 95% CI -2,9 do -2,7 kg), eksenatyd raz w tygodniu (-2,8 kg; 95% CI - 5,2 do -0,3 kg) lub liraglutyd (-2,2 kg; 95 % CI –3,5 do –0,9 kg), wykazały istotny spadek masy ciała. Dodatkowa analiza wykazała, że agoniści GLP-1 poprawiają skurczowe i rozkurczowe ciśnienie krwi, poziom cholesterolu i kontrolę glikemii. Autorzy doszli do wniosku, że agoniści GLP-1 stosowani u pacjentów otyłych, z cukrzycą lub bez, prowadzą do klinicznie istotnych korzyści w postaci utraty masy ciała. Można również zaobserwować dodatkowy pozytywny wpływ na ciśnienie krwi i poziom cholesterolu całkowitego.
Efektywność taranabanta - odwrotny agonista receptorów kannabinoidowych CB 1, który zmniejsza apetyt i zwiększa wydatek energetyczny, badano u otyłych pacjentów. Randomizowane badanie kliniczne wykazało zależną od dawki redukcję masy ciała w ciągu 12 tygodni przyjmowania leku. Opublikowano dane z czterech badań klinicznych III fazy, z których w dwóch oceniano stosunek korzyści do ryzyka stosowania leku w małych i wysokich dawkach, a w jednym oceniano skuteczność terapii u chorych na cukrzycę typu 2. Po 1 roku terapii taranabantem w dawce 0,5; 1 i 2 mg nastąpił spadek masy ciała średnio o 5,0; Odpowiednio 5,2 i 6,4 kg w porównaniu z 1,4 kg w grupie placebo (wszystkie p< 0,001) . Уменьшение массы тела на ≥ 5 и ≥ 10% достигнуто у большего количества пациентов в группах активной терапии по сравнению с плацебо (р < 0,001 для всех доз). Частота побочных эффектов в группах таранабанта была выше, чем в группе плацебо . В двойном слепом рандомизированном плацебо-контролируемом исследовании применялись более высокие дозы (2, 4 и 6 мг) в течение 104 нед. На основе анализа польза/риск терапия в дозе 6 мг была остановлена в течение 1-го года (пациенты с дозы 6 мг были переведены на 2 мг или плацебо) и в дозе 4 мг - в течение 2-го года (доза снижена с 4 до 2 мг). На 52 нед терапии среднее уменьшение массы тела составило 2,6; 6,6 и 8,1 кг соответственно в группах плацебо и таранабанта в дозе 2 и 4 мг (обе дозы р < 0,001 по сравнению с плацебо). У лиц, полностью завершивших 2-летний период лечения, изменения массы тела по сравнению с исходными данными были следующими: –1,4; –6,4 и –7,6 кг соответственно в группах плацебо и таранабанта в дозе 2 и 4 мг (обе дозы р < 0,001 по сравнению с плацебо). Продемонстрировано, что побочные эффекты значительно повышаются с увеличением дозы, особенно психические нарушения (депрессия, депрессивное настроение, тревога, суицидальные мысли, гнев и агрессия) . Таким образом, данные III фазы исследования показали, что и эффективность, и побочные эффекты ассоциировались с дозированием препарата, причем высокие дозы были более эффективны, но и побочных эффектов было больше. Эти данные послужили основой для прекращения клинических испытаний таранабанта для лечения ожирения.
Lorcaserin- potężny i selektywny agonista receptorów serotoninowych 5-HT 2C o szeregu właściwościach podobnych do fenfluraminy, działających na receptory serotoninowe 5-HT 2B i kojarzonych z wadami serca. Badania kliniczne wykazały znaczną skuteczność lorkaseryny w zmniejszaniu masy ciała w porównaniu z placebo, a także dobry profil bezpieczeństwa. W dwóch badaniach klinicznych III fazy, BLOOM (modyfikacja behawioralna i Lorcaserin for Overweight and Obesity Management) i BLOSSOM (modyfikacja behawioralna i Lorcaserin Second Study for Obesity Management), 6380 pacjentów z BMI w zakresie 27–45 kg/m2 zostało losowo przydzielonych do grupy otrzymującej 10 mg lorkaseryny 2 razy dziennie lub placebo. Czas trwania badania wynosił 52 tygodnie. Większą redukcję masy ciała wykazano w przypadku terapii lorkaseryną w porównaniu z placebo. Analiza zbiorczych danych z badań wykazała, że po 52 tygodniach terapii nastąpił spadek masy ciała o 5,8% w grupie stosującej lorkaserynę i o 2,5% w grupie placebo (p.< 0,0001) . В исследовании BLOOM среднее уменьшение массы тела через 1 год терапии составило 5,8 ± 0,2 кг в основной группе и 2,2 ± 0,1 кг в группе плацебо (p < 0,001) и удерживалось в течение 2 лет у 67,9% пациентов основной группы и 50,3% группы плацебо (р < 0,001). Уменьшение массы тела достигало более 5% от исходного уровня у 47,1 и 22,6% пациентов основной группы и плацебо соответственно . Наиболее частыми побочными эффектами были головная боль, головокружение и тошнота, существенно не отличавшиеся между группами.
W 2010 roku lek został odrzucony przez FDA ze względów bezpieczeństwa, w szczególności podczas eksperymentów na modelach zwierzęcych rejestrowano nowotwory. Jednak według nowych danych ryzyko zachorowania na raka u osób stosujących ten lek jest znikome. Jednak ryzyko rozwoju nadciśnienia i innych działań niepożądanych ze strony układu sercowo-naczyniowego pozostaje. W maju 2012 r. panel FDA zalecił zatwierdzenie lorkaseryny, leku odchudzającego, pomimo ciągłych obaw dotyczących bezpieczeństwa leku dla układu sercowo-naczyniowego. Jeśli FDA zatwierdzi lek, będzie to pierwszy nowy lek odchudzający dostępny na rynku amerykańskim od 10 lat.
Tesofenzyna- inhibitor wychwytu zwrotnego neuronów mediatorów serotoniny, dopaminy i noradrenaliny w strukturach mózgu odpowiedzialnych za apetyt. Jego działanie osiąga się poprzez tłumienie uczucia głodu i szybkiej sytości podczas jedzenia. Tesofenzyna została pierwotnie opracowana do leczenia chorób Parkinsona i Alzheimera, ale badanie wykazało zdolność leku do zmniejszania masy ciała. Co więcej, efekt ten był zależny od dawki. Zatem średnia zmiana masy ciała u pacjentów otyłych podczas 14-tygodniowej terapii tesofenzyną w dawce 0,125; 0,25; 0,5 i 1,0 mg wynosiło 2,1; 8,2; Odpowiednio 14,1 i 20,9%. Ogółem u 32,1% pacjentów doświadczyło co najmniej 5% zmniejszenie masy ciała podczas leczenia tesofenzyną (p< 0,001 в дозе 0,25; 0,5 и 1,0 мг по сравнению с плацебо). Из нежелательных явлений отмечено увеличение ЧСС с повышением дозы препарата. Изменений АД в основной группе не выявлено. Еще одним доказательством было рандомизированное двойное слепое плацебо-контролируемое исследование , включавшее 203 пациента с ожирением, средняя масса тела которых составляла чуть более 100 кг. Всем больным назначалась диета, а также прием тезофензина в одной из 3-х дозирований либо плацебо. Период наблюдения составил 6 мес. У пациентов, принимавших тезофензин в дозе 0,25; 0,5 и 1,0 мг, отмечалось уменьшение массы тела соответственно на 6,7; 11,3 и 12,8 кг, что достоверно превышало данные группы плацебо (p < 0,0001). Доля больных, достигших уменьшения массы тела на ≥ 5 кг составила 59, 87 и 91% на фоне терапии тезофензином в дозе 0,25; 0,5 и 1,0 мг соответственно по сравнению с 29% в контрольной группе. Применение тезофензина сопровождалось существенным увеличением ЧСС во всех исследуемых группах, а в группе лиц, получавших наиболее высокую дозу препарата, - повышением АД и высокой частотой изменения настроения . В связи с этим в III фазе клинических испытаний, согласованной с FDA, изучается эффективность и безопасность тезофензина в двух дозах 0,5 и 0,25 мг.
cetilistat- inhibitor lipazy trzustkowej – enzymu rozkładającego trójglicerydy w jelicie. Inaktywowane enzymy nie są w stanie hydrolizować trójglicerydów tłuszczów znajdujących się w diecie do wchłanialnych wolnych kwasów tłuszczowych i monoglicerydów. Lek ten jest podobny do orlistatu zatwierdzonego przez FDA, ale ze względu na inną strukturę molekularną oczekuje się, że będzie lepiej tolerowany i będzie miał mniej skutków ubocznych. Badania kliniczne I fazy u pacjentów otyłych zakończono w 2006 roku. Leczenie cetilistatem w 3 dawkach (60, 120 i 240 mg) przez 12 tygodni spowodowało istotnie większą utratę masy ciała niż w grupie placebo. Ponadto odsetek pacjentów, którzy osiągnęli co najmniej 5% redukcję początkowej masy ciała, był większy we wszystkich 3 grupach leczenia niż w grupie placebo. W badaniu klinicznym II fazy wzięło udział 612 otyłych pacjentów chorych na cukrzycę, losowo przydzielonych do grupy otrzymującej cetilistat lub placebo. Czas trwania badania wynosił 12 tygodni. Wykazano, że leczenie cetilistatem w dawce 80 i 120 mg prowadziło do istotnego zmniejszenia masy ciała w porównaniu z placebo (odpowiednio 3,85 i 4,32 wobec 2,86 kg). Jednocześnie spadek masy ciała był zbliżony do efektów terapii orlistatem (3,78 kg). Cetilistat był dobrze tolerowany i rzadziej zdarzało się odstawiać lek z powodu działań niepożądanych. Zatem liczba działań niepożądanych ze strony przewodu pokarmowego wyniosła 12% w przypadku orlistatu i 1–3% w przypadku cetylistatu. Jednak przyczyny tych różnic są niejasne. Obecnie w Japonii trwają badania kliniczne fazy III nad celestatem.
Naltrekson- długo działający antagonista receptorów opioidowych, wykazujący duże powinowactwo do tych ostatnich. Lek stosowany jest w leczeniu uzależnienia od opioidów i alkoholu, jednakże w grupie leczonej w trakcie terapii odnotowano zmniejszenie spożycia pokarmów, co doprowadziło do zmniejszenia masy ciała. Zakłada się, że receptory opioidowe w ośrodkowym układzie nerwowym są powiązane z aktywacją zachowań żywieniowych. Ustalono eksperymentalnie, że podawanie naloksonu szczurom prowadzi do krótkotrwałego zmniejszenia spożycia pokarmu poprzez blokadę beta-endorfin. W badaniach klinicznych z zastosowaniem naltreksonu (analogu naloksonu) zaobserwowano nierówny wpływ na utratę masy ciała u osób z nadwagą i niedowagą.
Terapia skojarzona
Połączenie bupropionu i naltreksonu(oba leki o przedłużonym uwalnianiu substancji czynnej (o przedłużonym uwalnianiu - SR) - lek Contrave). Lek ten wprowadzono po odkryciu, że naltrekson blokuje za pośrednictwem b-endorfiny hamowanie proopiomelanokortyny (POMC), prohormonu wspierającego wydzielanie hormonu stymulującego a-melanocyty (a-MSH), podczas gdy bupropion (poprzez receptory dopaminy) aktywuje POMC neurony i zwiększa wydzielanie anoreksogennego neuropeptydu a-MSH w podwzgórzu. Połączenie bupropionu i naltreksonu oddziałuje na procesy motywacji do jedzenia (efekt dopaminowy) oraz przyjemność/smakowitość jedzenia (efekt opioidowy). Badanie kliniczne oceniające skuteczność różnych dawek połączenia naltrekson/bupropion wykazało, że zwiększenie dawki naltreksonu nie prowadziło do większej utraty masy ciała. Jednakże, w ciągu 24 tygodni terapii, spadek masy ciała utrzymywał się. Wykazano, że skojarzona terapia naltreksonem SR/bupropionem SR przez 24 tygodnie spowodowała istotną poprawę w zakresie objawów depresyjnych, utratę masy ciała i poprawę kontroli żywieniowej u kobiet z nadwagą i otyłością z depresją.
Do randomizowanego, kontrolowanego badania COR-1 (Contrave Obesity Research 1) włączono 1742 pacjentów z BMI w zakresie 30–45 kg/m2 i łagodną otyłością lub BMI w zakresie 27–45 kg/m2 oraz wysokim poziomem cholesterolu LDL lub wysokim ciśnieniem krwi. Zgodnie z planem badania pacjentom przepisano niskokaloryczną dietę i ćwiczenia fizyczne oraz 1 z 3 schematów leczenia:
1) naltrekson o przedłużonym uwalnianiu substancji czynnej SR w dawce 32 mg/dobę + bupropion SR w dawce 360 mg/dobę w jednej tabletce ze stałymi dawkami leków (grupa NB32);
2) naltrekson SR w dawce 16 mg/dobę + bupropion SR 360 mg/dobę w jednej tabletce ze stałymi dawkami leków (grupa NB16);
3) grupa placebo.
Czas trwania badania wynosił 56 tygodni. Średnia masa ciała pacjentów przed badaniem wynosiła około 100 kg (220 funtów). Po leczeniu odnotowano spadek masy ciała o 1,4 kg w grupie placebo, o 4,9 kg w grupie NB16 i o 6,1 kg w grupie NB32. Odsetek pacjentów, którzy osiągnęli utratę masy ciała o 5% lub więcej, był również inny w każdej grupie: 48% w grupie NB32, 39% w grupie NB16 i 16% w grupie placebo. Większość pacjentów w grupie NB32 (25%) i NB16 (20%) straciła ponad 10% masy ciała w porównaniu z grupą placebo (7%). Jednocześnie zmniejszenie masy ciała z 5 do 10% przyczyniło się do lepszej kontroli poziomu glukozy w osoczu krwi, obniżenia poziomu cholesterolu w osoczu krwi i ryzyka rozwoju nadciśnienia tętniczego.
Do badania COR-diabetes, kontrolowanego placebo, prowadzonego metodą podwójnie ślepej próby, włączono 505 pacjentów z nadwagą lub otyłością i cukrzycą typu 2 (poziom HbA1C od 7 do 10%, średnio 8%), losowo przydzielonych do leczenia skojarzonego naltreksonem SR 32 mg/bupropionem SR 360 mg lub placebo. Czas trwania badania wynosił 56 tygodni. W grupie leczonej skojarzeniem naltrekson/bupropion zaobserwowano znaczną redukcję masy ciała (5 vs. 1,8%, p.< 0,001). При этом уменьшение массы тела ≥ 5% отмечено у 44,5% пациентов в основной группе по сравнению с 18,9% в группе плацебо. Показано улучшение контроля гликемии в группе комбинированной терапии по сравнению с группой плацебо. При этом целевой уровень HbA1C < 7% достигнут более чем у 44% пациентов в группе комбинированной терапии по сравнению с 26% в группе плацебо (p < 0,001). Авторы заключили, что в целом изучаемая комбинация препаратов хорошо переносилась. Наиболее частым побочным эффектом была тошнота.
Jednak w lutym 2011 roku FDA zawiesiła sprzedaż leku, tłumacząc tę decyzję koniecznością dodatkowych badań nad jego skutkami ubocznymi.
Połączenie bupropionu i zonisamidu. Skojarzenie bupropionu z lekiem przeciwpadaczkowym zonisamidem oceniano w trzech badaniach klinicznych II fazy. Zonisamid ma wieloskładnikowy mechanizm działania: działa hamująco na kanały sodowe bramkowane napięciem i kanały wapniowe typu T, wzmaga uwalnianie kwasu gamma-aminomasłowego i hamuje uwalnianie glutaminianu. Podczas leczenia zonisamidem występuje tendencja do utraty masy ciała. W randomizowanym badaniu klinicznym pacjenci otrzymujący terapię skojarzoną bupropionem i zonisamidem przez 24 tygodnie doświadczyli większej utraty masy ciała (9,2%) w porównaniu z pacjentami w grupach otrzymujących bupropion, zonisamid i placebo w monoterapii (6,6%, 3,6% i 0,4). odpowiednio %). Podobne wyniki uzyskano w randomizowanym, otwartym badaniu. Najczęstszymi działaniami niepożądanymi były bóle głowy, nudności i bezsenność. W tym samym okresie obserwacji zaobserwowano większy spadek masy ciała podczas terapii skojarzonej bupropionem zzonisamidem niż w przypadku terapii skojarzonej bupropionem i naltreksonem.
Terapia skojarzona z fenterminą i fenfluraminą. Trwające 28 tygodni randomizowane badanie kliniczne oceniające skuteczność tego połączenia u pacjentów otyłych wykazało istotny spadek masy ciała w porównaniu z placebo (15,5 vs. 4,9%, p.< 0,001). Однако в 1997 г. фенфлюрамин был отозван с рынка США в связи с появлением данных о формировании легочной гипертензии и клапанных пороков сердца на фоне применения указанным препаратом .
Połączenie fenterminy i topiramatu. Topiramat jest agonistą kwasu gamma-aminomasłowego i lekiem przeciwpadaczkowym testowanym w monoterapii w celu zmniejszenia masy ciała. Uważa się, że zmniejszenie apetytu spowodowane przez ten lek jest związane z receptorami glutaminianu podtypu kainianowego/AMPK, kanałami sodowymi bramkowanymi napięciem i aktywnością kwasu gamma-aminomasłowego. Jednakże dokładny mechanizm działania topiramatu na utratę masy ciała nie jest znany. W wielu randomizowanych badaniach klinicznych wykazano, że w porównaniu z placebo, monoterapia topiramatem powodowała znaczną redukcję masy ciała, którą obserwowano przez cały okres badania. Obawy dotyczące działań niepożądanych ze strony OUN i obwodowego układu nerwowego doprowadziły do rozpoczęcia badań III fazy topiramatu, które przerwano ze względu na dużą częstość występowania działań niepożądanych. Nie potwierdzono założeń dotyczących lepszej tolerancji topiramatu o przedłużonym uwalnianiu.
Istnieją dowody na to, że połączenie topiramatu o kontrolowanym uwalnianiu i fenterminy w małych dawkach jest skuteczne w leczeniu otyłości. W randomizowanym badaniu klinicznym 28-tygodniowa terapia skojarzeniem fenterminy i topiramatu spowodowała zmniejszenie masy ciała o 9,2% w porównaniu z 6,4% w przypadku samego topiramatu, fenterminy i placebo; Odpowiednio 6,1 i 1,7%. Ocena tolerancji i bezpieczeństwa terapii skojarzonej (badania EQUATE, EQUIP, CONQUER) pozwoliła ustalić takie skutki uboczne, jak przyspieszenie akcji serca, zaburzenia psychiczne (depresja, myśli samobójcze, problemy z pamięcią i koncentracją), a także wady wrodzone. Na podstawie danych dotyczących tolerancji i bezpieczeństwa jesienią 2010 roku FDA odrzuciła zatwierdzenie połączenia psychostymulującej fenterminy i przeciwdrgawkowego topiramatu (Qnexa).
Kombinacje pramlintydu. Neurohormonalna kontrola masy ciała obejmuje złożoną interakcję między leptyną i amyliną. Ustalono doświadczalnie, że u otyłych gryzoni leczeniu amyliną i leptyną towarzyszył znaczny spadek masy ciała na skutek zmniejszenia masy tkanki tłuszczowej. Dane te posłużyły jako podstawa do opracowania nowego leku złożonego, obejmującego pramlintyd (analog naturalnego hormonu amyliny) i metreleptynę (analog ludzkiego hormonu leptyny), syntetyzowany przez adipocyty, ważny regulator metabolizmu energetycznego zaangażowany w kontrola masy ciała. Małe badania kliniczne wykazały, że terapia skojarzona pramlintydem i metreleptyną była skuteczniejsza w zmniejszaniu masy ciała niż monoterapia jednym i drugim. Zatem po 20 tygodniach leczenia zmniejszenie masy ciała podczas terapii skojarzonej pramlintydem i mereleptyną wyniosło 12,7 ± 0,9% w porównaniu z 8,4 ± 0,9% w grupie pramlintydu (p< 0,001) и 8,2 ± 1,3% в группе метрелептина (p < 0,01) . Поэтому комбинацию прамлинтида с метрелептином стали рассматривать как новый интегрированный нейрогормональный подход в фармакотерапии ожирения. Однако в августе 2011 г. на этапе II фазы клинических испытаний было объявлено о прекращении испытаний комбинированного препарата прамлинтид/метрелептин для лечения ожирения.
Istnieją dane kliniczne oceniające działanie pramlintydu w skojarzeniu z sibutraminą i fenterminą. W otwartym badaniu, w którym uczestniczyli pacjenci otyli, spadek masy ciała podczas 24-tygodniowej terapii skojarzonej pramlintydu z sybutraminą wyniósł 11,1 ± 1,1%, w grupie otrzymującej pramlintyd z fenterminą – 11,3 ± 0,9%, pramlintyd w monoterapii – 3,7 ± 0,7% i 2,2 ± 0,7% w grupie placebo (p< 0,001). Общими побочными эффектами комбинированной терапии были тошнота и учащенное сердцебиение. Отмечено значительное повышение ЧСС и АД на фоне комбинированной терапии прамлинтид/сибутрамин (3,1 ± 1,2 уд./мин, p < 0,05; 2,7 ± 0,9 мм рт. ст., p < 0,01) и прамлинтид/фентермин (4,5 ± 1,3 уд./мин, p < 0,01; 3,5 ± 1,2 мм рт. ст., p < 0,001). В настоящее время изучается эффективность комбинированной терапии прамлинтидом с агонистом рецепторов ГПП-1 - эксенатидом для лечения ожирения у пациентов с СД и без этого заболевания .
Wniosek
Podobnie jak wiele innych problemów zdrowotnych na poziomie krajowym i światowym, profilaktyka i leczenie otyłości są dalekie od rozwiązania, o czym decyduje zarówno wiele czynników i uwarunkowań początkowych, jak i wieloskładnikowy charakter samego zadania. Podstawą leczenia jest ścisłe przestrzeganie stylu życia, zwiększona aktywność fizyczna i zmiana nawyków żywieniowych. Chociaż głównymi środkami farmakoterapeutycznymi stosowanymi w leczeniu otyłości są leki anoreksogenne, należy pamiętać, że mechanizm działania tych leków często prowadzi do potencjalnie niepożądanych skutków ubocznych. Planowanie terapii lekowej powinno opierać się na indywidualnej ocenie korzyści/ryzyka stosowania leków anoreksogennych przez pacjenta. Obecnie do długotrwałego leczenia otyłości dopuszczony jest wyłącznie orlistat. Jednocześnie priorytetem jest rozwój nowych leków, które wpływają na różne zaburzenia w układzie regulacji metabolizmu energetycznego i pozwalają nie tylko redukować masę ciała, ale także przeciwdziałać rozwojowi nawrotów choroby.
Spis referencji znajduje się w redakcji.
Historia blokerów receptorów opioidowych sięga połowy XX wieku, kiedy to zsyntetyzowano pierwsze allilowe pochodne pochodnych opium. W wyniku badań eksperymentalnych otrzymanych związków okazało się, że zastąpieniu grupy metylowej przy atomie azotu rdzenia morfinanu (rys. 1) masywniejszymi rodnikami węglowodorowymi towarzyszy znaczny wzrost powinowactwa (powinowactwa ) takiej zmodyfikowanej cząsteczki dla receptorów opioidowych w mózgu.
Ryż. 1. Podstawienie grupy metylowej (–CH 3) przy atomie azotu jądra morfinanu opioidów rodnikami węglowodorowymi (–R 3)
Wiadomo, że dla prawidłowego przejścia impulsu nerwowego przez synapsę konieczne jest, aby cząsteczki ligandów (substancji zdolnych do interakcji z odpowiednimi receptorami, w tym przypadku opioidami) po interakcji z receptorami błony postsynaptycznej zostały szybko usunięte ze szczeliny synaptycznej lub w niej enzymatycznie zniszczone, aby zwolnić miejsce dla kolejnej partii. Zwiększone powinowactwo tych modyfikowanych opioidów spowodowało, że dłużej niż zwykle pozostawały one na receptorach opioidowych, wolniej były usuwane ze szczeliny synaptycznej, a co za tym idzie, zakłócały przekazywanie impulsów nerwowych w synapsach endogennego układu opioidowego, które są związane z kontrolą percepcji bodźców bólowych i powstawaniem emocji. W ten sposób zsyntetyzowane związki nabyły właściwości antagonistów opium.
W procesie chemicznej przemiany „czystych” agonistów opium (narkotyków) stosuje się substancje o właściwościach pośrednich, tzw. agoniści-antagoniści, jak i „czyści” antagoniści opium, co należy uznać za przykład klasycznego przejścia ilości (powinowactwa do receptorów) w jakość (agonizm-antagonizm, ryc. 2). Co więcej, jedynie „czyści” antagoniści opiatów, czyli nalokson i naltrekson, można słusznie nazwać blokerami receptorów opioidowych.

Ryż. 2. Zmiany właściwości opioidów (z agonistycznych na antagonistyczne) w miarę wzrostu ich powinowactwa do receptorów opioidowych
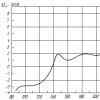
Przez pół wieku klinicznego stosowania blokerów receptorów opioidowych światowa społeczność medyczna przeszła wszystkie etapy dobrze znanej ewolucji poglądów na temat każdego nowego leku: „entuzjazm → rozczarowanie → zasłużone miejsce w codziennej praktyce”. Najbardziej wyraźny obraz tej ewolucji daje dynamika liczby publikacji poświęconych blokerom receptorów opioidowych w czasopismach medycznych na całym świecie (ryc. 3), według systemu informacyjnego Narodowej Biblioteki Medycznej Stanów Zjednoczonych Stany Ameryki „MedLine”.

Ryż. 3. Dynamika liczby publikacji zawierających w tytule słowo „nalokson” lub „naltrekson” według wyników wyszukiwania w systemie informacyjnym MedLine
Wyraźnie widać, że szczyt zainteresowania naloksonem przypada na lata 80. XX wieku, natomiast liczba publikacji poświęconych badaniu naltreksonu stale rośnie na przestrzeni ostatnich trzydziestu pięciu lat, tj. od momentu jego synteza do teraźniejszości.
Obszary klinicznego zastosowania naloksonu i naltreksonu wyznaczane są przez charakterystykę ich farmakokinetyki.
Nalokson jest lekiem krótko działającym. Jego okres półtrwania z organizmu mierzony jest w kilkudziesięciu minutach, a ze względu na intensywną biotransformację w wątrobie, lek przyjmowany doustnie jest nieskuteczny, dlatego przepisywany jest wyłącznie pozajelitowo (domięśniowo lub dożylnie).
W przeciwieństwie do naloksonu, naltrekson jest skuteczny po podaniu doustnym. Przy tej metodzie podawania działanie rozpoczyna się w ciągu 1–2 godzin i utrzymuje się do 24–48 godzin. Tak długotrwały efekt przy podaniu doustnym wynika z faktu, że główny metabolit naltreksonu – 6-beta-naltreksol – również ma właściwości blokera receptorów opioidowych, a jego okres półtrwania (około 13 godzin) wynosi około 3 razy dłuższy niż okres półtrwania samego naltreksonu (około 4 godziny).
Z uwagi na te okoliczności nalokson stosowany jest głównie w leczeniu ostrych zatruć opioidami, a naltrekson w ramach kompleksowej terapii przeciwnawrotowej schorzeń uzależnieniowych. Jednak ta sekcja „stref wpływów”, jak zostanie pokazane poniżej, jest bardzo warunkowa.
Nalokson w diagnostyce i leczeniu stanów uzależnienia od opioidów
Leczenie ostrych zatruć to nie jedyny obszar zastosowania naloksonu. Na przykład nalokson stosuje się do określenia obecności i nasilenia uzależnienia od opioidów. Ze względu na możliwość wystąpienia bolesnych dla pacjenta objawów odstawiennych, w tym wskazaniu stosuje się nalokson, głównie w przypadku pacjenta nieprzytomnego. W takim przypadku cele diagnostyczne (określenie rodzaju substancji psychoaktywnej, która spowodowała śpiączkę) i cele terapeutyczne (opieka w nagłych przypadkach) przepisywania tego leku można osiągnąć jednocześnie. Dodatkowo przed rozpoczęciem długotrwałego leczenia przeciwnawrotowego naltreksonem wykonuje się test naloksonowy.
Nalokson w dawce 0,2–0,4 mg podaje się dożylnie przez 5 minut, podskórnie lub domięśniowo, po czym pacjent jest uważnie obserwowany, starając się wykryć wczesne objawy zespół odstawienia: rozszerzone źrenice, przyspieszony oddech, łzawienie, wyciek z nosa i pocenie się. Jeżeli w ciągu 15–30 minut nie ma odpowiedzi na nalokson, lek podaje się ponownie dożylnie w dawce 0,4 mg lub podskórnie w dawce 0,4–0,8 mg i ponownie monitoruje się stan pacjenta. Brak reakcji na nalokson po wielokrotnym podaniu oznacza brak fizycznego uzależnienia od opioidów w momencie przeprowadzania tego badania. Należy pamiętać, że próba naloksonowa może dać wynik negatywny, także u pacjentów uzależnionych od opiatów, którzy są w remisji.
Konwencjonalne (doustne) postacie dawkowania naltreksonu w praktyce klinicznej
Naltrekson jest jednym z niewielu leków, które pierwotnie stworzono do leczenia uzależnienia od opioidów. Lek charakteryzuje się dużym powinowactwem do receptorów µ-opioidowych, przyjmowany jest doustnie i nie posiada właściwości psychoaktywnych, co minimalizuje ryzyko nadużywania.
Leczenie uzależnienia od opioidów naltreksonem rozpoczyna się natychmiast po zakończeniu detoksykacji i wykonaniu testu naloksonowego (patrz wyżej) w dawkach 50 mg – doustnie codziennie, 100 mg – doustnie co drugi dzień lub 150 mg – doustnie co 2 dni dni.
Naltrekson jest dobrze tolerowany przez pacjentów. W literaturze odnotowuje się jego zdolność do umiarkowanego działania stymulującego u pacjentów uzależnionych od opioidów oraz korzystnego wpływu na zaburzenia w zakresie abulicznym i astenicznym. Jednak u części pacjentów w okresie adaptacji do naltreksonu wzrasta poziom lęku i wzrasta drażliwość. Pojawienie się reakcji dysforycznych po wielokrotnym podaniu naloksonu uważa się za wskaźnik niepełnego leczenia zespołu odstawiennego. Częstość występowania innych działań niepożądanych (nudności, ból brzucha i głowy) jest stosunkowo niska. Najpoważniejszym działaniem niepożądanym jest hepatotoksyczne działanie naltreksonu, które obserwuje się jednak tylko przy stosowaniu bardzo dużych dawek (1400–2100 mg na tydzień).
Należy zauważyć, że naltrekson został oficjalnie zatwierdzony w Stanach Zjednoczonych jako lek na uzależnienie od opium w oparciu o dane dotyczące jego właściwości farmakologicznych i przy braku bezpośrednich przekonujących dowodów na jego skuteczność kliniczną. Głównym problemem ograniczającym skuteczność naltreksonu jest niska przestrzegalność zaleceń i wysoki odsetek nawrotów po zaprzestaniu jego stosowania. Stosowanie nagrody specjalnej za przyjmowanie leku, stosowanie naltreksonu w połączeniu z psychoterapią i poradnictwem, terapią rodzinną, a także w sytuacjach kontroli nad pacjentem przez organy ścigania (zwolnienie warunkowe) znacząco zwiększyło przestrzeganie i skuteczność leczenia terapii naltreksonem.
Jednakże rola naltreksonu w leczeniu stanów uzależnień nie ogranicza się do uzależnienia od opioidów. Pomimo tego, że etanol w odróżnieniu od opioidów sam nie oddziałuje z receptorami opioidowymi, jego stosowanie w poradniach leczenia uzależnień od alkoholu jest uzasadnione patogenetycznie. Dość duża liczba badań eksperymentalnych wykazała, że endogenny układ opioidowy jest ściśle powiązany z układem dopaminergicznym i wraz z nim bierze bezpośredni udział w powstawaniu uzależnienia od alkoholu. Najważniejsze jest to, że kiedy alkohol dostanie się do organizmu, pod wpływem dehydrogenazy alkoholowej, rozkłada się na szereg metabolitów, z których głównym jest aldehyd octowy. Jednocześnie alkohol powoduje uwolnienie z magazynu wolnej dopaminy. W wyniku kondensacji aldehydu octowego i dopaminy powstaje szereg endogennych związków o budowie niepeptydowej: tetrahydropapaweralina, salsolinol, tetrahydro-β-karbolina. Te produkty kondensacji alkoholu i dopaminy mogą oddziaływać z receptorami opioidowymi w mózgu i dlatego wykazują właściwości podobne do morfiny. Według współczesnych wyobrażeń właśnie dlatego naltrekson, jako bloker receptorów opioidowych, zmniejsza euforyczne i wzmacniające działanie etanolu.
Przeprowadzono znaczną liczbę podwójnie ślepych, randomizowanych, kontrolowanych placebo badań klinicznych dotyczących skuteczności leku w uzależnieniu od alkoholu, których dane są niejednoznaczne. Pewna niespójność wyników wiąże się ze stosunkowo krótkim okresem badań (12 tygodni). Jednak nawet dłuższe (choć niewielkie) badania nie dają wystarczającej jasności co do ostatecznego rozwiązania problemu. Większość autorów nadal odnotowuje nieznaczny wzrost przypadków całkowitej abstynencji w trakcie leczenia odwykowego lub w każdym razie spadek liczby nawrotów po załamaniach (jednorazowe spożycie alkoholu), a także spadek ilości spożywanego alkoholu u osób, które pij dalej. Wyniki przytoczonych badań wskazują na znaczenie łączenia terapii naltreksonem z regularnym poradnictwem dla pacjentów oraz z psychoterapią poznawczo-behawioralną (lub inną). Oprócz szeroko rozpowszechnionej „amerykańskiej metody” ciągłego leczenia naltreksonem istnieje metoda fińskich naukowców oparta na teorii ekstynkcji I. P. Pavlova. Według tej techniki pacjent przyjmuje naltrekson zaledwie na kilka godzin przed planowanym spożyciem alkoholu. Autorzy uważają, że w tych przypadkach naltrekson blokuje euforyczne działanie alkoholu i stopniowo „gaśnie” jego wzmacniające działanie, co z kolei zmniejsza częstotliwość spożywania alkoholu i jego ilość w trakcie spożycia. Poza sytuacjami związanymi ze spożyciem alkoholu pacjent zgodnie z tą techniką nie przyjmuje naltreksonu.
Podobnie jak w leczeniu uzależnienia od opiatów, naltrekson przepisywany jest doustnie raz dziennie osobom uzależnionym od alkoholu w dawce 50 mg (choć niektóre dane sugerują konieczność przyjmowania co najmniej 100 mg dziennie). Istnieją dowody na to, że w leczeniu uzależnienia od alkoholu naltrekson jest skuteczny tylko wtedy, gdy poziom przestrzegania zaleceń wynosi co najmniej 70–90% (za 100% zgodności uznaje się codzienne stosowanie). Ponadto niedawno wykazano, że lek ten jest najskuteczniejszy u pacjentów z pewnym podtypem receptorów opiatowych μ, co określa się na podstawie analizy genetycznej alleli odpowiednich genów kodujących ten receptor.
Obecnie prowadzone są badania nad tzw. terapią skojarzoną (np. farmakoterapią i psychoterapią behawioralną) uzależnienia od alkoholu, co pozwala doprecyzować możliwości farmakoterapii w ogóle, a także różnych kombinacji leków. W związku z tym w podwójnie ślepym, randomizowanym badaniu kontrolowanym placebo, które trwało 4 lata i obejmowało 1383 pacjentów uzależnionych od alkoholu, zbadano skuteczność naltreksonu, akamprozatu, standardowej terapii poznawczo-behawioralnej (CBT) i ich kombinacji. Wykazano, że naltrekson jest skuteczniejszy niż placebo jedynie w przypadku braku terapii poznawczo-behawioralnej. W przypadku CBT wszystkie pozostałe leki i ich kombinacje nie różniły się od siebie skutecznością, najwyraźniej ze względu na fakt, że CBT „przyćmiła” działanie leków. Jednocześnie naltrekson zmniejszał ryzyko nadużywania alkoholu w porównaniu z placebo, zmniejszał patologiczny głód alkoholu, a także zmniejszał liczbę dni intensywnego picia. Jakość życia pacjentów otrzymujących CBT była lepsza niż w przypadku wszystkich innych opcji leczenia.
Istnieją dowody na to, że naltrekson może być również skuteczny w leczeniu innych uzależnień, mianowicie uzależnienia od nikotyny i uzależnienia od kwasu γ-hydroksymasłowego (GHB). Wiadomo również, że naltrekson przynosi ulgę osobom cierpiącym na kleptomanię.
Należy zaznaczyć, że potencjalne zastosowanie naltreksonu nie ogranicza się do stanów uzależnienia. Aktywnie badane są możliwości leczenia naltreksonem dewiacyjnej hiperseksualności u nastolatków, stwardnienia rozsianego i autyzmu.
Długotrwały naltrekson jako sposób rozwiązywania problemów związanych z przestrzeganiem zaleceń. Wiwitrol
Jak już wspomniano, główną przyczyną spadku skuteczności leczenia stanów uzależnień naltreksonem jest niska przestrzegalność zaleceń i wysoki odsetek nawrotów choroby po odstawieniu leku. Trzy niezależne badania przeprowadzone odpowiednio w latach 2000, 2001 i 2002 (ryc. 4) wykazały, że liczba pacjentów przyjmujących przepisany doustnie naltrekson szybko spadała w czasie. Co więcej, około 50% całkowitej liczby pacjentów, którym przepisano naltrekson, nigdy nie odnawia recepty, mimo że lek jest dostarczany bezpłatnie.

Ryż. 4. Dynamika pacjentów odstawiających naltrekson w tabletkach na przykładzie osób uzależnionych od alkoholu [cyt. przez Harrisa KM i in. // Usługi psychiatryczne. - 2004. - Cz. 55. - s. 221]
Jedną z metod zwiększania przestrzegania zaleceń jest stosowanie naltreksonu w formie depot, która nie wymaga codziennego podawania leku.
Przykładem takiej formy depot jest naltrekson w postaci tabletek do implantacji (Prodetoxone), produkowany w Rosji, będący połączeniem naltreksonu w dawce 1000 mg i triamcynolonu w celu zapobiegania stanom zapalnym w miejscu implantacji.
Jednakże implantacja stałej postaci dawkowania jest, choć niewielką operacją, wymagającą odpowiednich warunków i wykwalifikowanego personelu.
Płynne postacie leków depot, które umożliwiają konwencjonalne podawanie w postaci zastrzyków, są znacznie wygodniejsze w obsłudze.
Obecnie najpowszechniejszą formą naltreksonu do wstrzykiwań jest lek Vivitrol. Jedna fiolka z wiwitrolem zawiera 380 mg naltreksonu w postaci mikrosfer (o średnicy około 100 mikronów). Mikrosfery to polimerowa matryca polilaktydo-glikolidowa (PLG) z osadzonym w niej aktywnym lekiem, który powoli rozpuszcza się w tkance mięśniowej. Po podaniu preparatu Vivitrol naltrekson uwalnia się z mikrosfer, osiągając maksymalne stężenie w ciągu 3 dni (Rysunek 5). Następnie, w wyniku dyfuzji i resorpcji matrycy polimerowej, naltrekson uwalnia się przez ponad 30 dni.

Ryż. 5. Dynamika stężenia naltreksonu w osoczu krwi pacjenta przy podawaniu doustnym i przy podawaniu Vivitrolu [cit. według Dunbara J. L. i in. // Alkoholizm, badania kliniczne i eksperymentalne. - 2006. - Cz. 30, nr 3. - s. 480–490]
Ze względu na to, że stężenie leku w osoczu krwi pozostaje przez długi czas prawie stałe, działanie farmakologiczne Vivitrolu znacznie różni się od działania doustnej formy naltreksonu. Skuteczność leku w leczeniu uzależnienia od alkoholu w osiąganiu i stabilizowaniu remisji wykazano w randomizowanych badaniach prowadzonych metodą podwójnie ślepej próby. Wykazano, że po sześciu miesiącach łączonej terapii psychospołecznej z Vivitrolem liczba dni „pijanych” zmniejszyła się 22,8 razy w porównaniu do poziomu początkowego i była o 90% mniejsza niż w przypadku skojarzonej terapii psychospołecznej z placebo (ryc. 6).

Ryż. 6. Liczba dni „pijanych” przed rozpoczęciem terapii i po sześciu miesiącach terapii psychospołecznej placebo i Vivitrolem [cyt. według O'Malley S. S. i in. // Dziennik Psychofarmakologii Klinicznej. - 2007. - Cz. 279, nr 5. - s. 507–512]
Notatka:
* - prowadzono terapię psychospołeczną według modelu BRENDA.
Wygodny schemat stosowania Vivitrolu to raz na 4 tygodnie. - pomaga rozwiązać problem zgodności. Według wyników specjalnego badania, 60% pacjentów było w stanie pomyślnie ukończyć 24-tygodniowy (168 dni) program leczenia Vivitrolem. Zatem stopień przestrzegania zaleceń dotyczących leczenia preparatem Vivitrol jest o rząd wielkości wyższy niż odpowiedni wskaźnik leczenia doustnym naltreksonem (patrz ryc. 4).
Badania wykazały, że Vivitrol jest ogólnie dobrze tolerowany. W przeciwieństwie do doustnego naltreksonu, nie opisano żadnego toksycznego działania Vivitrolu na wątrobę, co prawdopodobnie wynika z mniejszej syntezy pochodnych, w tym 6-beta-naltreksolu, w wyniku zmniejszonego metabolizmu pierwszego przejścia w wątrobie, a także fakt, że całkowita miesięczna dawka leku przy przepisywaniu Vivitrolu (380 mg) jest prawie 4 razy mniejsza niż przy przepisywaniu doustnego naltreksonu (50 mg/dzień × 30 dni = 1500 mg). Dlatego też Vivitrol można stosować u pacjentów z łagodną i średnio nasiloną dysfunkcją wątroby (klasa A i B w skali Child-Pugh). Najczęstszymi działaniami niepożądanymi w badaniach klinicznych były nudności, reakcje miejscowe i ból głowy. Ponieważ metabolizm Vivitrolu zachodzi bez udziału cytochromu P-450, nie przewiduje się wpływu induktorów i inhibitorów układu cytochromów na metabolizm Vivitrolu, co znacznie zmniejsza ryzyko interakcji z innymi lekami.
Analiza źródeł przytoczonych w tym przeglądzie wskazuje, że proces tworzenia nowych blokerów receptorów opioidowych i opracowywania nowych postaci ich dawkowania rozwijał się w kierunku zwiększania selektywności, zmniejszania liczby i nasilenia skutków ubocznych, a także wydłużania czasu trwania ich stosowania. działanie i łatwość użycia. Głównym celem tych działań było zwiększenie udziału pacjentów uzależnionych od substancji psychoaktywnych (głównie opioidów i alkoholu) w terapii, a w efekcie poprawa jakości życia tej kategorii pacjentów.
Biorąc pod uwagę całość jego właściwości, lek Vivitrol należy dziś uznać za szczyt tej ewolucji.
Literatura
- Blumberg H., Dayton H. B., Wolf P. S. Przeciwdziałanie narkotycznym antagonistom leków przeciwbólowych za pomocą narkotycznego antagonisty Naloksonu // Proceedings of the Society for Experimental Biology and Medicine. - 1966. - Cz. 123, nr 3. – s. 755–758.
- Foldes F. F., Davidson G. M., Duncalf D., Kuwabara S., Siker E. S. Wpływ na układ oddechowy, układ krążenia i działanie przeciwbólowe mieszanin naloksonu i narkotyków u znieczulonych osób // Dziennik kanadyjskiego stowarzyszenia anestezjologów. - 1965. - Cz. 12, nr 6. - s. 608–621.
- Jasinski D. R., Martin W. R., Haertzen C. A. Farmakologia człowieka i potencjał nadużywania N-allilonoroksymorfonu (naloksonu) // The Journal of Pharmacology and Experimental Therapeutics. - 1967. - Cz. 157, nr 2. – s. 420–426.
- Osterlitz H. W., Watt A. J. Parametry kinetyczne agonistów i antagonistów narkotyków, ze szczególnym uwzględnieniem N-allilonoroksymorfonu (naloksonu) // British Journal of Pharmacology and Chemotherapy. - 1968. - Cz. 33, nr 2. – s. 266–276.
- Smits S. E., Takemori A. E. Badania ilościowe dotyczące antagonizmu naloksonu niektórych narkotycznych i narkotycznych leków przeciwbólowych // British Journal of Pharmacology. - 1970. - Cz. 39, nr 3. – s. 627–638.
- Takemori A. E., Kupferberg H. J., Miller J. W. Ilościowe badania antagonizmu morfiny przez nalorfinę i nalokson // The Journal of Pharmacology and Experimental Therapeutics. - 1969. - Cz. 169, nr 1. - s. 39–45.
- http://www.ncbi.nlm.nih.gov.
- Minko A. I., Linsky I. V. Narkologia. - wyd. 2, wyd. i dodatkowe - M.: Eksmo, 2004. - 736 s.
- Krupitsky E. M., Ilyuk R. D., Eryshev O. F., Tsoi-Podosenin M. V. Nowoczesne farmakologiczne metody stabilizacji remisji i zapobiegania nawrotom uzależnienia od narkotyków // Przegląd Psychiatrii i Psychologii Medycznej im. V. M. Bekhtereva. - 2009. - nr 1. - s. 12–28.
- Lee M. C. i in. Czas zajmowania receptorów opioidowych przez naltrekson // Journal of Nuclear Medicine. - 1988. - Cz. 29. - s. 1207–1211.
- O'Brien C. P., Greenstein R., Mintz J., Woody G. E. Doświadczenia kliniczne z naltreksonem // American Journal of Drug and Alcohol Abuse. - 1975. - Cz. 2. - s. 365–377.
- Sivolap Yu. P., Savchenkov V. A., Yanushkevich M. V., Vandysh M. V. Ocena roli różnych klas leków w leczeniu uzależnienia od opioidów [Zasoby elektroniczne] // Psychiatria i psychofarmakologia. - 2004. - T. 6, nr 3. - Tryb dostępu: http://www.consilium-medicum.com.
- Litvintsev S. V. Organizacja pomocy w leczeniu uzależnień w Siłach Zbrojnych Federacji Rosyjskiej na obecnym etapie // Zagadnienia narkologii. - 2002. - nr 1. - s. 3–7.
- O'Brien C. P. Najnowsze osiągnięcia w farmakoterapii uzależnień // Journal of Consulting and Clinical Psychology. - 1996. - Cz. 64, nr 4. – s. 677–686.
- Kleber H.D., Kosten T.R. Indukcja naltreksonem: strategie psychologiczne i farmakologiczne // Journal of Clinical Psychiatry. - 1984. - Cz. 45. - s. 29–38.
- Grabowski J. Wpływ płatności warunkowej na przestrzeganie schematu naltreksonu // American Journal of Drug and Alcohol Abuse. - 1979. - Cz. 6, nr 3. – s. 355–365.
- Meyer RE i in. Bodziec heroinowy. - Nowy Jork, 1979. - R. 23–38, 93–118, 215–245.
- Callahan E. J., Rawson RA, McCleave B. i in. Leczenie uzależnienia od heroiny: sam naltrekson i terapia behawioralna // International Journal of Addiction. - 1980. - Cz. 15, nr 6. – s. 795–807.
- Cornish J. W., Metzger D., Woody G. E. i in. Farmakoterapia naltreksonem dla federalnych stażystów uzależnionych od opioidów // Journal of Substance Abuse Treatment. - 1997. - Cz. 14, nr 6. – s. 529–534.
- Trennant F. S., Rawson R. A., Cohen A. I., Mann A. Doświadczenia kliniczne z naltreksonem u osób uzależnionych od opioidów z przedmieść // Journal of Clinical Psychiatry. - 1984. - Cz. 45, nr 9. - s. 42–45.
- Anokhina I. P. Podstawowe mechanizmy biologiczne uzależnienia od alkoholu i narkotyków // Przewodnik po narkologii. - M., 2002. - T. 1. - s. 33–41.
- Anton R. F., Swift R. M. Aktualne framakoterapie alkoholizmu: perspektywa amerykańska // American Journal of Addiction. - 2003. - Cz. 12. - s. 53–68.
- O'Malley S. S., Jaffe A. J., Chang G. i in. Naltrekson i terapia umiejętności kopiowania w przypadku uzależnienia od alkoholu: badanie kontrolowane // Psychiatria ogólna. - 1992. - Cz. 49. - s. 881–887.
- Volpicelli J. R., Alterman AI, Hayashida M., O'Brien C. P. Naltrekson w leczeniu uzależnienia od alkoholu // Psychiatria ogólna. - 1992. - Cz. 49. - s. 876–880.
- Anton R. F., O'Malley S. S., Ciraulo D. A. i in. Połączone farmakoterapie i interwencje behawioralne w przypadku uzależnienia od alkoholu: badanie COMBINE: randomizowane badanie kontrolowane // JAMA. - 2006. - Cz. 295. - s. 2003–2017.
- Carmen B., Angeles M., Ana M., Maria A. J. Skuteczność i bezpieczeństwo naltreksonu i akamprozatu w leczeniu uzależnienia od alkoholu: przegląd systematyczny // Uzależnienie. - 2004. - Cz. 99. - s. 811–828.
- Sinclair J.D. Dowody na zastosowanie naltreksonu i różne sposoby jego stosowania w leczeniu alkoholizmu // Alkohol i alkoholizm. - 2001. - Cz. 36. - s. 2–10.
- Volpicelli J. R., Rhines K. C., Rhines K. C. i in. Uzależnienie od alkoholu naltreksonowego. Rola subiektywnej zgodności // Archives of Psychiatry. - 1997. - Cz. 54. - s. 737–742.
- Anton R., Oroshi G., O'Malley S. i in. Ocena receptora opioidowego (OPRM1) jako predyktor odpowiedzi naltreksonu na leczenie uzależnienia od alkoholu: wyniki badania połączonej farmakoterapii i interwencji behawioralnych w leczeniu uzależnienia od alkoholu (COMBINE) // Archives of Psychiatry. - 2008. - Cz. 65, nr 2. – s. 135–144.
- Byars J. A., Frost-Pineda K., Jacobs W. S., Gold M. S. Naltrekson nasila działanie nikotynowej terapii zastępczej u palących kobiet // Journal of Addictive Diseases. - 2005. - Cz. 24, nr 2. - s. 49–60.
- Caputo F., Vignoli T., Lorenzini F., Ciuffoli E., Re A. D., Stefanini. Tłumienie głodu kwasu gamma-hydroksymasłowego przez podawanie naltreksonu: trzy opisy przypadków // Neurofarmakologia kliniczna. - 2005. - Cz. 28, nr 2. - s. 87–89.
- Grant J. E. Badanie wyników pacjentów z kleptomanią leczonych naltreksonem: przegląd wykresów // Neurofarmakologia kliniczna. - 2005. - Cz. 28, nr 1. - s. 11–14.
- Ryback R.S. Naltrekson w leczeniu młodocianych przestępców seksualnych // Journal of Clinical Psychiatry. - 2004. - Cz. 65, nr 7. – s. 982–986.
- Agrawal Y.P. Terapia naltreksonem w małych dawkach w stwardnieniu rozsianym // Hipotezy medyczne. - 2005. - Cz. 64, nr 4. – s. 721–724.
- Remschmidta X. Autyzm. Objawy kliniczne, przyczyny i leczenie [Zasoby elektroniczne]. - Tryb dostępu: http://www.autism.ru/read.asp?id=151&vol=21.
- http://www.hippocrat.info/prodetokson.htm [Zasoby elektroniczne].
- Bartus R. T., Emerich D. F., Hotz J. i in. Vivitrex, postać naltreksonu do wstrzykiwań o przedłużonym uwalnianiu, dostarcza dowodów farmakokinetycznych i farmakodynamicznych na skuteczność przez 1 miesiąc u szczurów. // Neuropsychofarmakologia. - 2003. - Cz. 28. - s. 1973–1982.
- Johnson B. A., Ait-Daoud N., Aubin H. J. i in. Pilotażowa ocena bezpieczeństwa i tolerancji podawania dawek wielokrotnych długo działającego naltreksonu do wstrzykiwań (Vivitrex) u pacjentów uzależnionych od alkoholu // Alkoholizm, badania kliniczne i eksperymentalne. - 2004. - Cz. 28. - s. 1356–1361.
- Garbutt J. C., Kranzler H. R., O'Malley S. S. i in. Skuteczność i tolerancja długo działającego naltreksonu do wstrzykiwań w leczeniu uzależnienia od alkoholu: randomizowane badanie kontrolowane // JAMA. - 2005. - Cz. 293. - s. 1617–1625.
- Volpicelli J. R., Pettinati H. M., McLellan A. T., O'Brien C. P.Łączenie leków i psychospołecznego leczenia uzależnień. Podejście BRENDA. - Guilford Press, 2001. - 208 s.
8.1. Opioidowe (narkotyczne) leki przeciwbólowe i ich antagoniści
Działanie farmakologiczne opioidowych leków przeciwbólowych i ich antagonistów wynika z interakcji z receptorami opioidowymi, które występują zarówno w ośrodkowym układzie nerwowym, jak i tkankach obwodowych.
Opierając się na zasadzie interakcji leków przeciwbólowych z tej grupy z receptorami opioidowymi, można je przedstawić w postaci następujących grup.
Agoniści
Morfina Promedol Fentanyl Sufentanyl Agoniści-antagoniści i częściowi agoniści Pentazocyna Nalbufina Butorfanol Buprenorfina
Wiele opioidowych leków przeciwbólowych należy do pierwszej grupy substancji. Jednakże w tym charakterze można również stosować agonistów-antagonistów, jeśli dominują ich właściwości agonistyczne (na przykład pentazocyna), jak również częściowych agonistów. Ponieważ te leki przeciwbólowe oddziałują z receptorami opioidowymi, nazywane są opioidami.
Opioidowe leki przeciwbólowe mają wyraźny wpływ depresyjny na ośrodkowy układ nerwowy. Ma działanie przeciwbólowe, nasenne i przeciwkaszlowe. Ponadto większość z nich zmienia nastrój (występuje euforia) i powoduje uzależnienie od narkotyków (psychiczne i fizyczne).
Do grupy opioidowych leków przeciwbólowych zalicza się szereg leków otrzymywanych zarówno z surowców roślinnych, jak i syntetycznie.
1 Informacje na temat pochodzenia terminu „środek przeciwbólowy” można znaleźć w rozdziale 5.
Agoniści receptorów opioidowych
Alkaloid 1 morfina stała się powszechna w praktyce medycznej. Jest izolowany z opium 2, czyli zamrożonego mlecznego soku wypływającego z nacięć głów maku nasennego - Papawersomniferum (ryc. 8.2). Opium przeznaczone do celów medycznych musi zawierać co najmniej 10% morfiny. W sumie opium zawiera ponad 20 alkaloidów.
Ze względu na budowę chemiczną niektóre alkaloidy opium należą do pochodnych fenantrenu, inne zaś do pochodnych izochinoliny.
Pochodne fenantrenu (morfina, kodeina itp.) charakteryzują się głównie działaniem depresyjnym na ośrodkowy układ nerwowy (przeciwbólowe, przeciwkaszlowe), a alkaloidy izochinolinowe (papaweryna itp.) mają bezpośrednie działanie przeciwskurczowe na mięśnie gładkie.
W tej części, spośród alkaloidów opium, za typową przedstawicielkę opioidowych (narkotycznych) leków przeciwbólowych uznawana będzie jedynie morfina.
Głównym działaniem morfiny jest jej działanie przeciwbólowe. Morfina ma dość wyraźną selektywność działania przeciwbólowego. Inne rodzaje wrażliwości (dotyk, wrażliwość na temperaturę, słuch, wzrok)
nie) w dawkach terapeutycznych nie tłumi.
Mechanizm działania przeciwbólowego morfiny nie jest w pełni poznany. Niemniej jednak istnieją podstawy, aby sądzić, że składa się ona z następujących głównych elementów: 1) zahamowania procesu międzyneuronalnego przekazywania impulsów bólowych w centralnej części drogi aferentnej oraz 2) zaburzeń subiektywnej percepcji emocjonalnej, oceny bólu i bólu reakcja na to 3.
Mechanizm działania przeciwbólowego morfiny wynika z jej interakcji z receptorami opioidowymi (μ > κ ≈ δ), których jest agonistą. Pobudzenie receptorów opioidowych przez morfinę objawia się aktywacją endogennego układu antynocyceptywnego i zaburzeniem międzyneuronalnej transmisji bodźców bólowych na różnych poziomach ośrodkowego układu nerwowego. Zatem bezpośrednio

Ryż. 8.2. Tabletki nasenne - Papaver somniferum L. (zawiera alkaloidy morfinę, kodeinę, papawerynę itp.).
1 Znaczenie terminu „alkaloid” znajduje się w rozdziale 1.3.
2 Z języka greckiego. przeciw - sok. Opium pozyskiwane jest ręcznie poprzez odcięcie niedojrzałych główek maku, a następnie zebranie suszonego na powietrzu mlecznego soku.
3 W ostatnich latach pojawiły się dowody na to, że opioidy mają obwodowy składnik przeciwbólowy. Tym samym wykazano, że w eksperymencie prowadzonym w warunkach stanu zapalnego opioidy zmniejszają wrażliwość bólową na stres mechaniczny. Oczywiście procesy opioidergiczne biorą udział w modulowaniu bólu w tkankach objętych stanem zapalnym.

VA SERTURNERA (1783-1841). W 1806 roku z maku nasennego wyizolował alkaloid morfinę. Był to pierwszy alkaloid otrzymany w postaci oczyszczonej.
hamujący wpływ morfiny na neurony rdzenia kręgowego. W tym przypadku zaburzenie międzyneuronalnej transmisji pobudzenia następuje na poziomie rogów grzbietowych rdzenia kręgowego. Nie bez znaczenia jest także wpływ morfiny na jądra nadrdzeniowe, które biorą udział w zstępującej kontroli aktywności neuronów w rogach grzbietowych rdzenia kręgowego. Eksperyment wykazał, że wprowadzenie morfiny do niektórych z tych jąder (na przykład do istoty szarej okołoprzewodowej, do komórek siatkowatych i jąder komórek olbrzymich) powoduje działanie przeciwbólowe. O znaczeniu układu zstępującego świadczy także fakt, że zniszczenie dużego jądra szwu znacznie zmniejsza przeciwbólowe działanie morfiny. Zatem hamujący wpływ morfiny na przekazywanie impulsów bólowych w rdzeniu kręgowym z pierwotnych włókien doprowadzających do interneuronów polega na wzroście zstępujących wpływów hamujących i bezpośrednim działaniu hamującym na przekazywanie interneuronów w rdzeniu kręgowym. Tego typu działania zlokalizowane są zarówno na neuronach postsynaptycznych, jak i na poziomie zakończeń presynaptycznych. W tym drugim przypadku morfina stymulując presynaptyczne receptory opioidowe na zakończeniach pierwotnych włókien doprowadzających, zmniejsza uwalnianie mediatorów (np. glutaminianu, substancji P) biorących udział w przekazywaniu bodźców nocyceptywnych. Hamowanie neuronów postsynaptycznych wynika z ich hiperpolaryzacji (w wyniku aktywacji postsynaptycznych kanałów K +). Zakłócenie transmisji międzyneuronalnej w rdzeniu kręgowym przez morfinę zmniejsza intensywność impulsów docierających do wstępujących dróg doprowadzających, a także zmniejsza reakcje motoryczne i autonomiczne (ryc. 8.3).
Zmiana percepcji bólu najwyraźniej wiąże się nie tylko ze zmniejszeniem dopływu impulsów bólowych do leżących nad nimi odcinków, ale także z uspokajającym działaniem morfiny. To ostatnie w sposób oczywisty wpływa na ocenę bólu i jego konotacji emocjonalnej, co ma ważny na motoryczne i autonomiczne objawy bólu. Rola stanu psychicznego w ocenie bólu jest bardzo ważna. Wystarczy zauważyć, że pozytywny wpływ placebo na niektóre bóle sięga 35-40%.
Uspokajające działanie morfiny może wynikać z jej działania na neurony kory mózgowej, na aktywację wstępującego tworzenia siatkowatego pnia mózgu, a także na układ limbiczny i podwzgórze. Wiadomo np., że morfina hamuje reakcję aktywacyjną kory mózgowej (hamuje desynchronizację EEG na bodźce zewnętrzne), a także reakcję układu limbicznego i podwzgórza na impulsy aferentne.
Jednym z typowych przejawów psychotropowego działania morfiny jest stan, jaki ona powoduje euforia 1 , czyli podwyższony nastrój,
1 Z języka greckiego. ue - Cienki, fero - Wytrzymam to.

Ryż. 8.3. Możliwe punkty działania morfiny.
Działanie przeciwbólowe morfiny wynika z jej stymulującego działania na receptory opioidowe na różnych poziomach ośrodkowego układu nerwowego.
1 - wpływ na receptory presynaptyczne pierwotnych włókien aferentnych (prowadzi do zmniejszenia uwalniania mediatorów, np. substancji P, glutaminianu); 2 - wpływ na receptory postsynaptyczne neuronów w rogu grzbietowym rdzenia kręgowego, prowadząc do zahamowania ich aktywności; 3, 4 - aktywacja układu antynocyceptywnego śródmózgowia i rdzenia przedłużonego (centralna istota szara, jądra szwu) wzmaga zstępujące działanie hamujące na przewodzenie impulsów bólowych w rogach grzbietowych rdzenia kręgowego; 5 - hamowanie międzyneuronalnego przekazywania impulsów bólowych na poziomie wzgórza; 6 - podczas stanu zapalnego zmniejsza się wrażliwość zakończeń nerwów doprowadzających. PAG - istota szara okołoprzewodowa; LC – miejsce sinawe; NRM - duży rdzeń raphe; HA - włókna adrenergiczne; Enk. - włókna enkefalinergiczne; Serot. - włókna serotoninergiczne; minus - działanie hamujące.
poczucie komfortu psychicznego, pozytywne postrzeganie otoczenia i perspektyw życiowych, niezależnie od rzeczywistości. Euforia jest szczególnie wyraźna przy wielokrotnym stosowaniu morfiny. Jednak u niektórych osób występuje zjawisko odwrotne: zły stan zdrowia, negatywne emocje (dysforia 1).
W dawkach terapeutycznych morfina powoduje senność, a w sprzyjających warunkach sprzyja rozwojowi snu 2. Sen wywołany morfiną jest zwykle powierzchowny i łatwo przerywany przez bodźce zewnętrzne.
Jednym z przejawów ośrodkowego działania morfiny jest obniżenie temperatury ciała związane z hamowaniem ośrodka termoregulacji zlokalizowanego w podwzgórzu. Wyraźną hipotermię obserwuje się jednak dopiero po podaniu dużych dawek morfiny. Morfina może jednak działać stymulująco na niektóre ośrodki podwzgórza. W szczególności prowadzi to do zwiększenia uwalniania hormonu antydiuretycznego (wazopresyny) i zmniejszenia diurezy.
Zwężenie źrenic (zwężenie źrenic) obserwowane podczas podawania morfiny (zwłaszcza w dawkach toksycznych) również ma genezę centralną i wiąże się z pobudzeniem ośrodków nerwu okoruchowego. To ostatnie ma najwyraźniej charakter wtórny i powstaje w wyniku działania morfiny na leżące nad nim części ośrodkowego układu nerwowego. Założenie to opiera się na fakcie, że morfina nie powoduje zwężenia źrenic u obłuszczonych psów.
Znaczące miejsce w farmakodynamice morfiny zajmuje jej działanie na rdzeń przedłużony, a przede wszystkim na ośrodek oddechowy. Morfina (od dawek terapeutycznych) hamuje ośrodek oddechowy, zmniejszając jego pobudliwość na dwutlenek węgla i działanie odruchowe. Po pierwsze, następuje spadek częstotliwości oddechów, który jest kompensowany wzrostem ich amplitudy. Po zwiększeniu dawki do subtoksycznej rytm oddychania zmniejsza się jeszcze bardziej, zmniejsza się amplituda pojedynczych oddechów i objętość minutowa. Często obserwuje się nieprawidłowy rytm oddechowy, możliwe jest okresowe oddychanie (przy toksycznych dawkach substancji). W przypadku zatrucia morfiną śmierć następuje w wyniku paraliżu ośrodka oddechowego.
Morfina hamuje centralne elementy odruchu kaszlowego i ma wyraźne działanie przeciwkaszlowe.
Morfina z reguły działa przygnębiająco na ośrodek wymiotów. Jednak w niektórych przypadkach może powodować nudności i wymioty. Jest to związane ze stymulującym działaniem morfiny na chemoreceptory strefy spustowej (Strefa wyzwalania), znajduje się na dnie komory IV i aktywuje ośrodek wymiotów (patrz ryc. 15.3). Morfina pobudza ośrodek nerwu błędnego, zwłaszcza w dużych dawkach. Występuje bradykardia. Nie ma praktycznie żadnego wpływu na ośrodek naczynioruchowy. Odruchy rdzeniowe zwykle nie ulegają zmianie po podaniu morfiny w dawkach terapeutycznych, są tłumione.
Zatem wpływ morfiny na ośrodkowy układ nerwowy jest dość zróżnicowany (tabela 8.2).
Morfina wywiera wyraźny wpływ na wiele narządów mięśni gładkich zawierających receptory opioidowe. W przeciwieństwie do alkaloidów opium z serii izochinolin (na przykład papaweryny), morfina stymuluje mięśnie gładkie, zwiększając ich napięcie.
1 Z języka greckiego. dys - odmowa, fero - Wytrzymam to.
2 Morfina otrzymała swoją nazwę ze względu na działanie hipnotyczne (na cześć greckiego boga snów, Morfeusza).
Tabela 8.2. Główne skutki morfiny

Z przewodu żołądkowo-jelitowego obserwuje się wzrost napięcia zwieraczy i jelit, zmniejszenie ruchliwości jelit, co sprzyja przemieszczaniu się ich zawartości oraz wzrost segmentacji jelit. Ponadto zmniejsza się wydzielanie trzustki i wydzielanie żółci. Wszystko to spowalnia przepływ treści pokarmowej przez jelita. Sprzyja temu również intensywniejsze wchłanianie wody z jelita i zagęszczanie jego zawartości. W rezultacie rozwija się zaparcie (zaparcie).
Morfina może znacznie zwiększyć napięcie zwieracza Oddiego (zwieracza brodawki wątrobowo-trzustkowej) i dróg żółciowych, co zakłóca proces przedostawania się żółci do jelita. Zmniejsza się także wydzielanie soku trzustkowego.
Morfina zwiększa napięcie i aktywność skurczową moczowodów. Tonizuje również zwieracz pęcherza, co utrudnia oddawanie moczu.
Pod wpływem morfiny zwiększa się napięcie mięśni oskrzeli, co może wynikać zarówno z jej działania na mięśniowe receptory opioidowe, jak i z uwalniania histaminy.
Morfina praktycznie nie działa bezpośrednio na naczynia krwionośne.
W dawkach terapeutycznych zwykle nie powoduje zmiany poziomu ciśnienia krwi. Zwiększając dawkę może powodować lekkie niedociśnienie, które przypisuje się lekkiemu zahamowaniu ośrodka naczynioruchowego i uwalnianiu histaminy. W wyniku działania morfiny może rozwinąć się niedociśnienie ortostatyczne.
Morfina nie wchłania się wystarczająco dobrze z przewodu pokarmowego. Ponadto znaczna jego część jest w pierwszej fazie inaktywowana w wątrobie
przechodząc przez to. W związku z tym, aby uzyskać szybszy i wyraźniejszy efekt, lek zwykle podaje się pozajelitowo. Czas działania przeciwbólowego morfiny wynosi 4-6 godzin. Jest on zdeterminowany dość szybką biotransformacją morfiny w wątrobie i jej usuwaniem z organizmu 1 . Morfina słabo przenika przez barierę krew-mózg (około 1% podanej dawki przedostaje się do tkanki mózgowej). Morfina w postaci niezmienionej (10%) i jej koniugaty (90%) są wydalane głównie przez nerki oraz w małych ilościach (7-10%) przez przewód pokarmowy, gdzie przedostają się do żółci.
Omnopon (pantopon), będący mieszaniną chlorowodorków 5 alkaloidów opium, zarówno z serii fenantrenowej (morfina, kodeina, tebaina), jak i izochinolinowej (papaweryna, narkotyna), jest czasami stosowany jako jeden z substytutów morfiny. Farmakodynamika omnoponu jest na ogół podobna do farmakodynamiki morfiny. Jedną z różnic jest to, że Omnopon zwiększa napięcie mięśni gładkich w mniejszym stopniu niż morfina.
Oprócz morfiny w praktyce medycznej znalazło zastosowanie wiele leków syntetycznych i półsyntetycznych. Strukturę niektórych z nich podano poniżej.
Do leków przeciwbólowych zaliczają się pochodne piperydyny, które mają spektrum działania receptorowego podobne do morfiny (µ > κ ≈ δ; Tabela 8.3). Jednym z powszechnie stosowanych w praktyce leków z tej serii jest promedol (chlorowodorek trimeperydyny). Pod względem działania przeciwbólowego jest 2-4 razy gorszy od morfiny 2. Czas działania wynosi 3-4 godziny. Nudności i wymioty występują rzadziej niż w przypadku morfiny. Nieco mniej przygnębia ośrodek oddechowy.
Promedol (oraz przeciwbólowa meperydyna o podobnej budowie i działaniu) ulegają w organizmie biotransformacji, tworząc neurotoksyczny N-demetylowany metabolit. Ten ostatni stymuluje ośrodkowy układ nerwowy (możliwe drżenie, drżenie mięśni, wzmożenie odruchów, drgawki). Metabolit ma długi okres półtrwania (t 1/2 = 15-20 godzin). Dlatego promedol (i meperydyna) zalecany jest wyłącznie do krótkotrwałego stosowania (do 48 godzin).

1 Wyizolowano metabolit 6-glukuronid morfiny. Jest bardziej aktywna niż morfina i działa nieco dłużej.
2 Aby uzyskać pożądany efekt, promedol stosuje się w większych dawkach niż morfina.

Tabela 8.3. Wpływ opioidów na różne rodzaje receptory

Notatka. Plus - agoniści; plus w nawiasach - częściowi agoniści; minus - antagoniści.
Napięcie narządów mięśni gładkich zmniejsza się (moczowody, oskrzela) lub wzrasta (jelita, drogi żółciowe), ale ma gorsze działanie spazmogenne niż morfina. W niewielkim stopniu zwiększa aktywność skurczową mięśniówki macicy. Dobrze wchłania się z przewodu pokarmowego.
Inny przedstawiciel pochodnych piperydyny, fentanyl (sentonyl), wykazuje bardzo wysoką aktywność przeciwbólową. Według danych eksperymentalnych uzyskanych różnymi metodami badawczymi jest 100-400 razy bardziej aktywna niż morfina 1. Cechą charakterystyczną fentanylu jest krótki czas łagodzenia bólu (20-30 minut po podaniu dożylnym). Efekt pojawia się w ciągu 1-3 minut. Fentanyl powoduje wyraźną (aż do zatrzymania oddechu), ale krótkotrwałą depresję ośrodka oddechowego.
Zwiększa napięcie mięśni szkieletowych, w tym mięśni klatki piersiowej. Ta ostatnia upośledza wentylację płuc i utrudnia oddychanie sztuczne lub wspomagane. Aby zmniejszyć napięcie mięśniowe, zwykle stosuje się leki przeciwdepolaryzacyjne podobne do kurary. Często występuje bradykardia (można ją wyeliminować za pomocą atropiny). Jest metabolizowany w wątrobie. Jednak ustanie działania wynika głównie z redystrybucji fentanylu w organizmie (stężenie fentanylu w ośrodkowym układzie nerwowym zmniejsza się ze względu na wzrost jego zawartości w tkankach obwodowych).
Zsyntetyzowano jeszcze bardziej aktywne analogi fentanylu – cytrynian sufentanylu i alfentanyl. Właściwości farmakologiczne, w tym skutki uboczne obu leków są zasadniczo podobne do fentanylu. Jednak przy podawaniu pozajelitowym ich działanie następuje jeszcze szybciej niż fentanylu. W zależności od czasu trwania działania przeciwbólowego i „okresu półtrwania” (t 1 / 2) można je ułożyć w następującej kolejności: fentanyl (t 1 / 2 = 3,6 godziny) > sufentanyl (t 1 / 2 = 2,7 godziny) > alfentanyl (t 1 / 2 = 1,3 godziny). Zanik działania jest także szybszy w przypadku sufentanylu i alfentanylu. W przeciwieństwie do fentanylu i sufentanylu, alfentanyl ma bardziej typowe działanie hipotensyjne.
Należy pamiętać, że czas działania fentanylu i jego analogów zależy od wieku pacjenta (jest dłuższy u osób starszych) i czynności wątroby (efekt znacznie wzrasta w przypadku marskości wątroby).
U wszystkich agonistów receptorów opioidowych rozwija się uzależnienie (w tym sieciowanie) i uzależnienie od narkotyków (psychiczne i fizyczne).
Opioidowe leki przeciwbólowe stosuje się w przypadku uporczywego bólu związanego z urazami, przebytymi operacjami, zawałem mięśnia sercowego, nowotworami złośliwymi itp. Wiele z tych leków ma wyraźne działanie przeciwkaszlowe.
Fentanyl stosuje się głównie w połączeniu z lekiem przeciwpsychotycznym droperydolem (obie substancje wchodzą w skład leku wzgórzowego; synonim - Innovar) w leczeniu neuroleptanalgezji 2.
1 Fentanyl jest przepisywany w dawkach 100-krotnie lub więcej mniejszych niż dawka morfiny.
2 Neuroleptanalgezja jest szczególnym rodzajem znieczulenia ogólnego. Osiąga się to poprzez skojarzone stosowanie leków przeciwpsychotycznych (neuroleptyków), np. droperydolu (patrz rozdział 11; 11.1) i aktywnego opioidowego leku przeciwbólowego (grupa fentanylu). W tym przypadku działanie przeciwpsychotyczne (neuroleptyczne) łączy się z wyraźnym działaniem przeciwbólowym . Świadomość zostaje zachowana. Obydwa leki działają szybko i krótko. Ułatwia to wchodzenie i wychodzenie ze stanu neuroleptanalgezji. Jeśli do leków na neuroleptanalgezję dodaje się podtlenek azotu, ta metoda znieczulenia ogólnego nazywa się neuroleptanestezją. Ponadto jednym z rodzajów znieczulenia ogólnego stosowanym podczas operacji chirurgicznych jest tzw znieczulenie zrównoważone. Odnosi się to do jednoczesnego stosowania ultrakrótko działającego barbituranu, opioidowego leku przeciwbólowego, antydepolaryzującego środka zwiotczającego mięśnie i podtlenku azotu.
Opioidowe leki przeciwbólowe są szeroko stosowane w premedykacji przed interwencjami chirurgicznymi. Morfinę podaje się także w znieczuleniu miejscowym, gdyż wzmacnia działanie środków znieczulających miejscowo.
W ostatnich latach w leczeniu bólu przewlekłego z powodzeniem stosuje się transdermalny system fentanylu (plastry z fentanylem aplikuje się podskórnie co 72 godziny).
Stosując opioidowe leki przeciwbólowe (np. promedol) w celu łagodzenia bólu podczas porodu, należy wziąć pod uwagę, że wszystkie one przenikają przez barierę łożyskową i powodują depresję ośrodka oddechowego płodu. Jeśli pomimo środków ostrożności u noworodka wystąpi asfiksja, do żyły pępowinowej wstrzykuje się opioidowego antagonistę leku przeciwbólowego, nalokson.
W przypadku bólu spowodowanego skurczami dróg żółciowych lub moczowodów, a także wrzodów trawiennych żołądka i dwunastnicy, kolki jelitowej, bardziej wskazane jest stosowanie promedolu i omnoponu, ponieważ w mniejszym stopniu zwiększają napięcie mięśni gładkich niż morfina. Jednakże w takich przypadkach wskazane jest podawanie tych leków w połączeniu z m-blokerami (np. atropiną) lub miotropowymi lekami przeciwskurczowymi (np. papaweryną). Czasami opioidowe leki przeciwbólowe są przepisywane w przypadku ciężkiego kaszlu, a także duszności związanej z niewydolnością lewej komory.
Działania niepożądane mogą obejmować nudności, wymioty, bradykardię, zaparcia itp. Leki należy stosować ostrożnie u pacjentów z niewydolnością oddechową lub zaburzeniami czynności wątroby. Są przeciwwskazane u dzieci do 3 roku życia i w starszym wieku (ze względu na hamujący wpływ na ośrodek oddechowy).
Agoniści-antagoniści i częściowi agoniści receptorów opioidowych
Agoniści-antagoniści działają odmiennie na różne typy receptorów opioidowych: niektóre typy receptorów stymulują (działanie agonistyczne), inne blokują (działanie antagonistyczne). Do leków takich zalicza się pentazocyna, butorfanol, nalbufina (patrz tabele 8.3 i 8.4).
Tabela 8.4. Charakterystyka porównawcza opioidowych leków przeciwbólowych

Notatka. Liczba plusów wskazuje na nasilenie efektu; ? - znikomy wpływ.
Pierwszym lekiem tego typu wprowadzonym do praktyki lekarskiej była pentazocyna (Lexir, Fortral). W porównaniu do pochodnych fenantrenu, w strukturze pentazocyny brakuje jednego z pierścieni. Lek jest agonistą receptorów δ i κ oraz antagonistą receptorów μ. Jest gorszy od morfiny pod względem działania przeciwbólowego i czasu działania. Pentazocyna zwróciła na siebie uwagę ze względu na to, że jej stosowanie wiąże się ze stosunkowo niskim ryzykiem uzależnienia od narkotyków (w porównaniu z agonistami opioidowymi lekami przeciwbólowymi) (nie powoduje euforii, może powodować dysforię). Hamuje oddychanie w nieco mniejszym stopniu niż morfina, a podczas stosowania rzadziej pojawiają się zaparcia. Pentazocyna powoduje wzrost ciśnienia w tętnicy płucnej; Zwiększa się ośrodkowe ciśnienie żylne, co prowadzi do zwiększonego obciążenia wstępnego serca. Zwiększa pracę serca. Ze względu na działanie hemodynamiczne, pentazocyny nie należy stosować podczas zawału mięśnia sercowego. Dobrze wchłania się z przewodu pokarmowego. Pentazocyna jest również antagonistą agonistycznych opioidowych leków przeciwbólowych, ale jej działanie jest słabo wyrażone. Antagonizm objawia się w szczególności tym, że po podaniu pentazocyny osobom uzależnionym od leków przeciwbólowych będących agonistami opioidów rozwija się u nich zespół odstawienny.
Do agonistów-antagonistów zalicza się także butorfanol (Moradol, Stadol) i nalbufinę (Nubain).
Butorfanol ma właściwości farmakologiczne podobne do pentazocyny. Jest agonistą receptora κ i słabym antagonistą receptora μ. Jest 3-5 razy bardziej aktywna niż morfina. Podobnie jak pentazocyna zwiększa ciśnienie w tętnicy płucnej i wzmaga pracę serca, dlatego nie zaleca się jej stosowania w zawale mięśnia sercowego. Oddychanie jest mniej depresyjne niż morfina. Uzależnienie od narkotyków rzadziej jest spowodowane przez morfinę. Podawać dożylnie lub domięśniowo, czasami donosowo (po 3-4 godzinach).
Nalbufina jest agonistą receptora κ i słabym antagonistą receptora μ. Jego działanie jest w przybliżeniu takie samo jak morfiny. Farmakokinetyka jest podobna do farmakokinetyki morfiny. Nie ma praktycznie żadnego wpływu na hemodynamikę. Uzależnienie od narkotyków występuje rzadko (w przybliżeniu z taką samą częstością jak pentazocyna). Podawać pozajelitowo po 3-6 godzinach.
Buprenorfina (Buprenex) jest częściowym agonistą receptora μ. Jego działanie przeciwbólowe przewyższa 20-60 razy morfinę i działa dłużej (powoli odrywa się od połączenia z receptorami opioidowymi). Efekt rozwija się wolniej niż w przypadku morfiny. Mniejszy wpływ na przewód żołądkowo-jelitowy niż morfina. Nie zwiększa ciśnienia w pęcherzyku żółciowym i przewodzie trzustkowym. W mniejszym stopniu opóźnia przepływ treści pokarmowej przez jelita. Stosunkowo dobrze wchłania się z przewodu pokarmowego (patrz tabela 8.5). Główna część niezmienionego leku jest wydalana przez jelita, metabolity przez nerki. Potencjał narkogenny jest stosunkowo niski. Odstawienie jest mniej dotkliwe niż w przypadku morfiny.
Podawać pozajelitowo i podjęzykowo (po 6 godzinach). Po podaniu podjęzykowym biodostępność wynosi około 50%.
1 Różnice w działaniu przeciwbólowym występują przy różnych dawkach leków. Jednak w praktyce skuteczność przeciwbólowa substancji jest ważniejsza, gdy są stosowane w dawkach terapeutycznych. Okazuje się, że to drugie jest praktycznie takie samo dla wszystkich wymienionych w tabeli opioidowych leków przeciwbólowych. 8.4.
Przypadkowe lub celowe przedawkowanie opioidowych leków przeciwbólowych prowadzi do ostrego zatrucia. Objawia się oszołomieniem, utratą przytomności i śpiączką. Oddech jest przygnębiony. Minimalna objętość oddechowa stopniowo maleje. Pojawia się nieregularny i okresowy oddech. Skóra jest blada, zimna, błony śluzowe sinicze. Jednym z objawów ostrego zatrucia morfiną i podobnymi substancjami jest ciężkie zwężenie źrenic (jednak przy ciężkim niedotlenieniu źrenice rozszerzają się). Krążenie krwi jest zaburzone. Temperatura ciała spada. Śmierć następuje w wyniku paraliżu ośrodka oddechowego.
Tabela 8.5. Farmakokinetyka niektórych leków przeciwbólowych działających ośrodkowo

Uwaga: i/n – donosowo, i/v – dożylnie, i/m – domięśniowo, podskórnie – podskórnie, i.v
Leczenie ostrego zatrucia opioidowymi lekami przeciwbólowymi przebiega następująco. Przede wszystkim należy wykonać płukanie żołądka, a także wprowadzić adsorbenty i środki przeczyszczające zawierające sól fizjologiczną. Jest to szczególnie istotne w przypadku dojelitowego podawania substancji i ich niepełnego wchłaniania.
Kiedy rozwinie się efekt toksyczny, użyj określonego opioidowy antagonista leku przeciwbólowego Nalokson (Narcan), który blokuje wszystkie typy receptorów opioidowych. Nalokson nie ma właściwości agonisty receptorów opioidowych. Odwraca nie tylko depresję oddechową, ale także większość innych skutków opioidowych leków przeciwbólowych, w tym agonistów i antagonistów. W przypadku przedawkowania buprenorfiny nalokson jest znacznie mniej skuteczny. Po podaniu doustnym lek wchłania się, ale większość ulega zniszczeniu podczas przejścia przez wątrobę. Nalokson podaje się dożylnie i domięśniowo. Działanie następuje szybko (po około 1 minucie) i utrzymuje się do 2-4 godzin.
Stworzono również długo działającego (10 godzin) antagonistę, nalmefen, do podawania dożylnego.
W przypadku ostrego zatrucia opioidowymi lekami przeciwbólowymi może być konieczne zastosowanie sztucznego oddychania. Ze względu na obniżenie temperatury ciała, takim pacjentom należy zapewnić ciepło. Jeżeli śmierć w wyniku zatrucia opioidami metabolizowanymi głównie w organizmie, takimi jak morfina, nie zostanie
wprowadzone w ciągu pierwszych 6-12 godzin, rokowanie uważa się za korzystne, ponieważ w tym czasie większość podawanego leku jest inaktywowana.
Naltrekson jest także uniwersalnym opioidowym antagonistą leku przeciwbólowego. Jest około 2 razy bardziej aktywny niż nalokson i działa znacznie dłużej (24-48 godzin). Skutki uboczne mogą powodować bezsenność, nudności, kurczowy ból brzucha i ból stawów. Przeznaczony wyłącznie do stosowania dojelitowego. Stosowany jest głównie w leczeniu uzależnienia od opioidów.
Jak już wspomniano, przy długotrwałym stosowaniu opioidowych leków przeciwbólowych rozwija się uzależnienie (psychiczne i fizyczne 1), które jest zwykle przyczyną przewlekłego zatrucia tymi lekami.
Pojawienie się uzależnienia od narkotyków można w dużej mierze wytłumaczyć zdolnością opioidowych leków przeciwbólowych do wywoływania euforii. Jednocześnie eliminowane są nieprzyjemne emocje i zmęczenie, pojawia się dobry nastrój i pewność siebie, a zdolność do pracy zostaje częściowo przywrócona. Euforię zwykle zastępuje wrażliwy, łatwo przerywany sen.
Przy powtarzających się dawkach opioidowych leków przeciwbólowych rozwija się uzależnienie. Dlatego narkomani potrzebują coraz większych dawek odpowiednich substancji, aby osiągnąć euforię.
Nagłe zaprzestanie podawania leku, który spowodował uzależnienie, prowadzi do zjawiska deprywacji (abstynencji). Pojawia się strach, niepokój, melancholia i bezsenność. Możliwy jest niepokój, agresywność i inne objawy. Wiele funkcji fizjologicznych jest upośledzonych. Czasami następuje zapaść. W ciężkich przypadkach odstawienie może być śmiertelne. Podanie opioidowego leku przeciwbólowego łagodzi objawy deprywacji. Abstynencja występuje również wtedy, gdy na tle istniejącego uzależnienia od narkotyków pacjentowi podaje się nalokson (a także pentazocynę).
Stopniowo narasta przewlekłe zatrucie. Zmniejsza się wydajność psychiczna i fizyczna, obserwuje się wrażliwość skóry, wychudzenie, pragnienie, zaparcia, wypadanie włosów itp.
Leczenie uzależnienia od opioidowych leków przeciwbólowych jest zadaniem bardzo trudnym. Wymagane jest długotrwałe leczenie szpitalne. Stopniowo zmniejszaj dawkę i częstotliwość podawania opioidowego leku przeciwbólowego. Długo działające opioidowe leki przeciwbólowe podaje się z wolniejszym ustaniem działania (więcej informacji można znaleźć w podręcznikach i podręcznikach z zakresu narkologii i psychiatrii). Jednak radykalne wyleczenie obserwuje się w stosunkowo niewielkim odsetku przypadków. U większości pacjentów występują nawroty. W tym względzie bardzo ważne są środki zapobiegawcze: ścisła kontrola przechowywania, przepisywania i wydawania opioidowych leków przeciwbólowych.